1.晶格振动和晶体热学性质
黄昆的《固体物理学》的第五章:晶格振动和晶体热学性质 - 知乎 (zhihu.com)




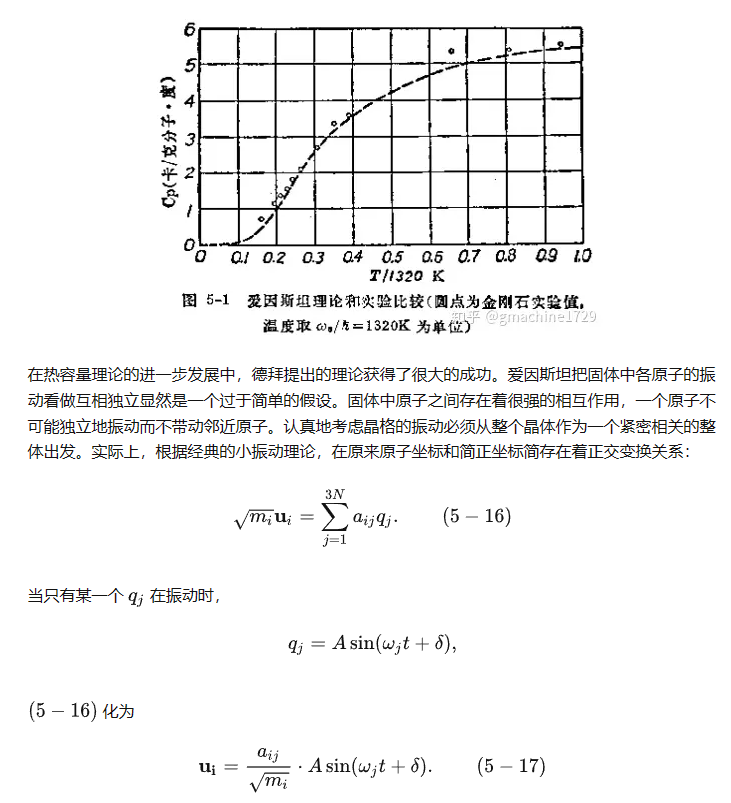






这里的简正坐标也可以看作简正原子的振动
简正坐标和坐标之间有变换关系
声子
2.简正坐标 声子
固体物理:2.3 谐振子能量 声子 - 知乎 (zhihu.com)
1. 广义坐标
1. 广义坐标就是在处理多物体的体系时,把整个体系看成一个物体,把多个物体(包括N自由度)的运动看成一个物体的不同部分在N维空间内的运动。但是区别于单个物体,一个体系物体内部的各部分的质量是不同的
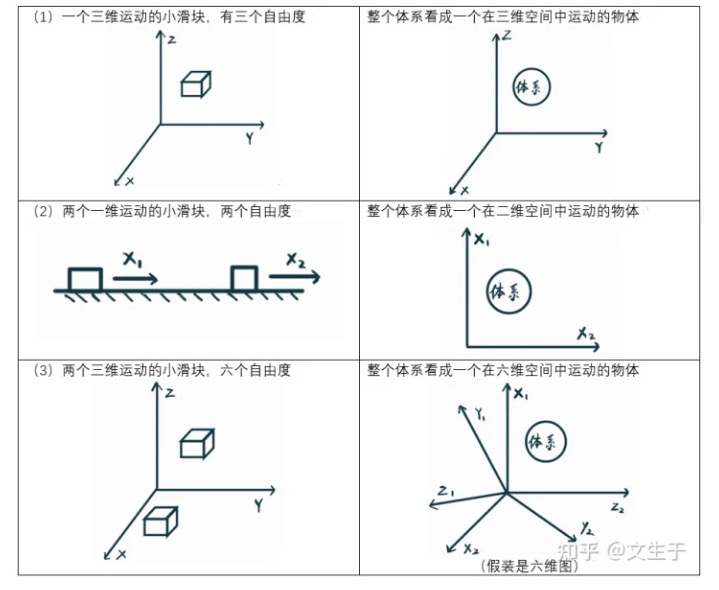
对于一个物体的能量,也可以按照自由度拆分为各个分能量

原子链中格波的表达式
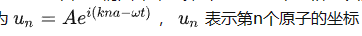
每个独立格波,都是一维单原子链的运动。体系的运动,就是多个格波的叠加
分运动的叠加是矢量叠加,需要给各个运动乘上对应方向的单位矢量
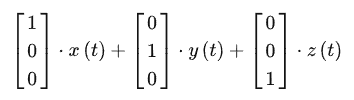
- 什么是坐标系:坐标系只不过是一种描述方式,只要找到两两不平行的一组向量,就能作为坐标系

其中,$k=m\frac{2\pi}{Na}, m=1-N$ 对应N个独立格波

这样,可以把一个格波中所有原子的坐标改写为(把振动作为基本坐标)

也就是只沿着第一个方向有分量。看成是整个个体的振动

2. 声子
==引入简正坐标就是为了引入不同简正模式的叠加==

经典理论中,谐振子的能量包括动能和势能

量子力学中,谐振子的能量为
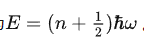
声子就是一个个的能量单元
3. 玻色爱因斯坦分布
晶体中的振动总能就是所有独立格波能量的累加,原子坐标就看成n个简正坐标的叠加


每个格波都对应着一定数量的声子
温度升高,所有格波的声子数都增加,温度下降,声子数减小,这也符合温度越大能量越高的判断
一个独立格波的平均能量就变成

T=0 K时,声子数趋近于零,但是格波的能量不为零,存在零点能 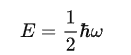
当温度很高时

这个时候一个格波的能量和温度T成正比
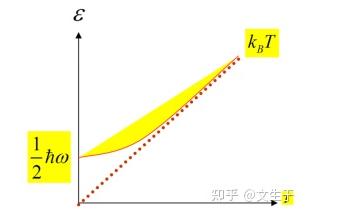
可以用来解决热容得问题,三维单原子链中有3N个独立格波
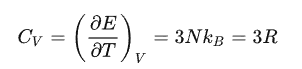
3. 态密度
独立格波的总数=总的自由度数=维数*原子数=原胞数*维数*单个原胞内的原子个数

绘制一张图,横坐标是频率,纵坐标是对应的能量
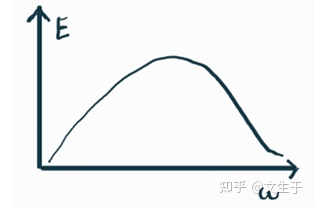
要计算总能,只需要对w积分,但是这里把无穷变为积分还需要打个补丁,考虑落在w-dw范围内的模式数是多少,也就是模式密度

给模式密度积分,就能得到独立格波总数

g(w)横坐标是w,纵坐标是不同的模式数
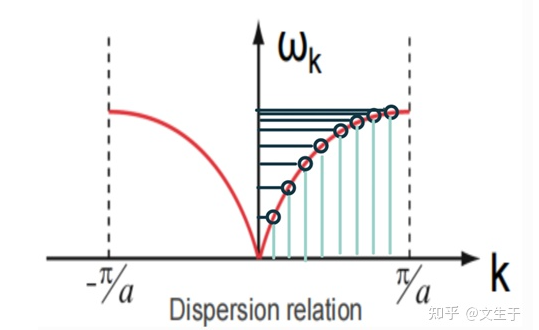
g(w)的值有些地方密集,有些地方分散每一处的模式密度都不同。模式密度处处不同,但是模式对应的波矢是均匀分布的。
==波矢空间的密度分布==
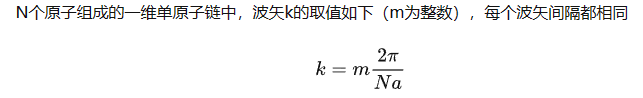
仿照g(w),可以定义一个单位波矢空间间隔内的振动模式数量,称为波矢密度

可以得到p(k)是一个常数
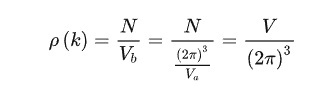
联系两个式子

例如

下面可以计算格波的总能量


色散关系很重要
4. 晶格热容
计算固体的热容,主要需要求声子的总能量。总能量等于各格波的能量叠加

确定态密度最关键的是知道色散关系
杜隆-波提

爱因斯坦模型直接定义有3N个谐振子(考虑了分布,有温度的效应)

德拜模型,考虑了线性的色散关系 w=cK (连续介质弹性波)
5. 固体物理 from simon
- 非简谐性质:非抛物线形状的势能曲线
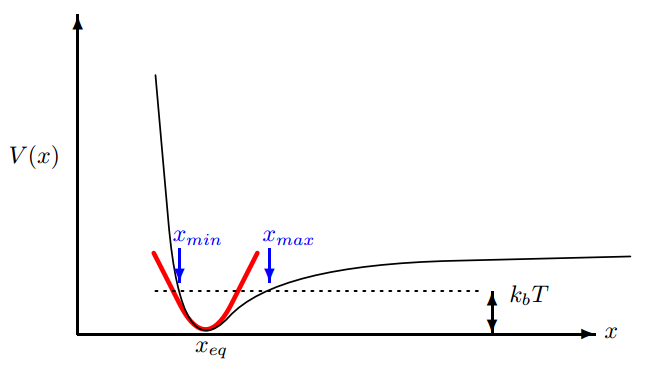
热膨胀:在温度升高时,x${max}$ 要大于x${min}$ ,导致原子间距离的平均值向右侧移动,晶格体积膨胀。平衡位置是能量最小值,但是不是原子振动的平均值(在小振动时由于是抛物线,所以相等,但是在高温下,不再是抛物线形状
==热膨胀本身对应着非简谐性==
6.色散关系
低频区对应着声学支,高频区对应着光学支(光学声子容易被光激发)。当波矢很小(即长波长)时,声子的频率通常与波矢成线性关系(通常叫做声波的“线性区域”),这是由于声子振动类似于经典的弹性波。
当波矢增大(即短波长)时,声子的频率趋于饱和,达到一个上限值,这通常与原子间的力常数以及晶体的弹性性质有关。
共有3N条色散关系,意味着每个k对应n个频率
声子态密度就是 g(w) ,如果一条w(k)是平的,就对应着这个能量区间有很多模式,那声子态密度就是一个大突出。
色散关系是本征的,可以根据色散关系对温度进行响应
能带也是本征的,根据相应电子色散进行响应
7. 为什么声子态密度可以分元素画出来
from matchat ai



[1] Chen J, et al. Real-Space Visualization of Frequency-Dependent Anisotropy of Atomic Vibrations. https://arxiv.org/abs/2312.01694
[2] Zhang L, et al. Multifield phonon spectrometrics of structured crystals and liquids. https://arxiv.org/abs/2001.02052
[3] Liu Y, et al. Normal modes in liquids and gases. https://arxiv.org/abs/2304.09778
[4] Wang H, et al. Algebraic theory of crystal vibrations: Singularities and zeros in vibrations of 1D and 2D lattices. https://arxiv.org/abs/1506.01707
[5] Smith R, et al. Density oscillations in multi-component molecular mixtures. https://arxiv.org/abs/0803.2950
[6] Li M, et al. Vibrational modes from MD simulation. https://arxiv.org/abs/2102.07957
[7] Zhao X, et al. Chapter 2: Normal mode decomposition of atomic motion in solids. https://arxiv.org/abs/2309.03140



8. 计算声子谱
见vasp使用教程
来源
固体中简正模式的解释
力常数矩阵

每个原子的位移可以分为多个简谐振动的和 从这里可以计算力常数矩阵进而求出每个q的动力学矩阵,进而求出每个q的频率w,维度为3n


动力学矩阵


有限位移法(Finite Displacement Method)是一种用于计算晶体声子谱的数值方法,其基本原理是通过对晶体结构中的原子进行微小的位移,计算相应的力,从而构建力常数矩阵,进而求解声子频率和振动模式。
具体步骤如下:
结构优化:
首先,对晶体结构进行高精度的优化,以获得平衡的原子位置和晶格常数。
在优化过程中,需要设置适当的收敛标准,如能量收敛(EDIFF)和力收敛(EDIFFG)等,以确保结构的准确性。
构建超胞:
在优化后的结构基础上,使用Phonopy等工具构建超胞。
超胞的尺寸通常是原胞的整数倍,具体尺寸取决于所需的声子计算精度和计算资源。
原子位移:
对超胞中的每个原子进行微小的位移,通常在0.01到0.03埃之间。
这些位移可以是沿着晶体轴的正负方向,或者在不同方向上进行。
力计算:
在每个位移结构上,进行单点能量计算,获取每个原子所受的力。
这些力数据用于构建力常数矩阵。
构建力常数矩阵:
利用获得的力数据,构建力常数矩阵。
该矩阵描述了原子间的相互作用,是计算声子谱的基础。
声子谱计算:
通过对力常数矩阵进行对角化,得到声子的频率和振动模式。
进一步,可以绘制声子色散曲线,分析声子的行为。
注意事项:
有限位移法的计算精度高度依赖于力的计算精度,因此在结构优化和单点计算中需要设置严格的收敛标准。
位移幅度的选择需要平衡计算精度和计算量,过大的位移可能导致力常数矩阵的非线性,过小的位移可能导致数值误差。
计算过程中,可能需要考虑晶体的对称性,以减少计算量。
通过上述步骤,有限位移法能够有效地计算晶体的声子谱,为研究材料的热力学性质、动力学稳定性等提供重要信息。
9. 热导

导热时才有声子的传播,就像声音传播一样,也是振动的传播。
光学支没有群速度(斜率),所以不参与热导
非谐和缺陷都会散射声子,降低热导
10 撒点和dos计算




展宽是计算delta函数时转换为高斯函数需要的
所以撒点越密集越好,能够尽可能准确地得到所有分布细节







四面体法通过四个顶点上的q值线性插值得到未插值点的信息
11 . 热椭球和均方根位移
只能用来考虑平衡态,不能考虑离子跃迁







零点振动能保证在T=0 MSD不为零,仍在振动
转载请注明来源 有问题可通过github提交issue