- 1.计算方法、流程与输入文件
- 1.关于结构优化
- 关于赝势的选择及赝势类型
- 2.测k点 /直接测KSPACING
- 3.ISMEAR与SIGMA
- 4.计算dos与band
- 5. 测试NCORE和KPAR
- 6.关于扩三倍胞
- 7. 构建POTCAR
- 9. HSE06 计算能带
- 9.自旋轨道耦合计算能带
- 10 .做异质结
- 11.做差分电荷密度
- 12. 如何考虑加U和加磁性
- 马德隆规则(n+l 规则)
- 失去电子时:
- 3d 与 4s 的能量高低
- 2. 失电子顺序
- 原子序数和轨道收缩
- 电子数和相互作用
- 关于轨道局域性
- 1. 轨道类型 (l) 对局域性的影响
- 2. 主量子数 (n) 对局域性的影响
- 综合判断
- 能带(Band Structure)
- 态密度(Density of States, DOS)
- 2. 第一性原理计算
- 1. 为什么不考虑加 U?
- 2. 为什么不考虑加磁性?
- 1. 强烈推荐加 U 的情况 (误差破坏性大)
- 2. 通常不加 U 的情况 (误差破坏性小)
- 核心区别:定性错误 vs. 定量错误
- 1. NiO (d⁸, 未满): U 决定了“能不能动” (局域化)
- 2. ZnO (d¹⁰, 全满): U 决定了“有多深” (能级位置)
- 1. 能量位置的巨大差异 (物理现实)
- 2. VASP 赝势的处理 (计算现实)
- 重要
- 重要
- 关于LASPH和LMAXMIX
- 13. 磁性
- 14. 如何设置参数加快计算速度
- 15. 关于vasp的输出中的free energy TOTEN 和energy without entropy 和 sigma >>0
- 12. 计算声子谱
- 2. 关于并行参数
- 试试ISYM = 0
- 1. internal ERROR RSPHER:running out of buffer
- 2 . 报错 Error EDDDAV: Call to ZHEGV failed. Returncode =
- 3. 报错 ERROR FEXCP: supplied Exchange-correletion table is too small, maximal index :
- 4. ERROR FEXCP: supplied Exchange-correletion table is too small, maximal index : 3885
- 5. Sub-Space-Matrix is not hermitian
- 6. HNFORM: ERROR: k-point generating vectors and reciprocal lattice are incommensurate.
- 7.自洽不收敛
- 8. 内存溢出/BAD TERMINATION OF ONE OF YOUR APPLICATION PROCESSES
1.计算方法、流程与输入文件
1.关于结构优化
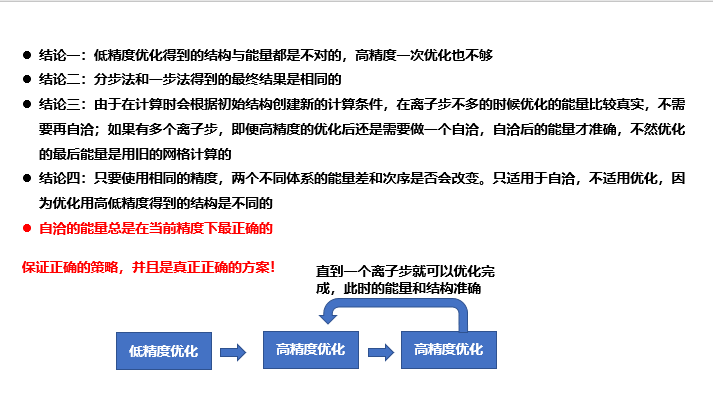
为什么vasp优化的能量不能直接拿来用而需要再做一步自洽?因为优化过程中晶格在变化,但是优化过程中的G点网格没有更新,所以最后用的G点网格不适用最后的晶格常数
QE最后会有一步自洽,所以QE的能量是准的
ABACUS优化的能量是不准的
使用收敛的参数不会出现优化后的结构拿去自洽后能量变化的问题,也不会有优化后的结构再拿去cp CONTCAR POSCAR后优化还可以继续优化的问题,所以归根结底是收敛的参数的问题
用于验证的目录:五洲
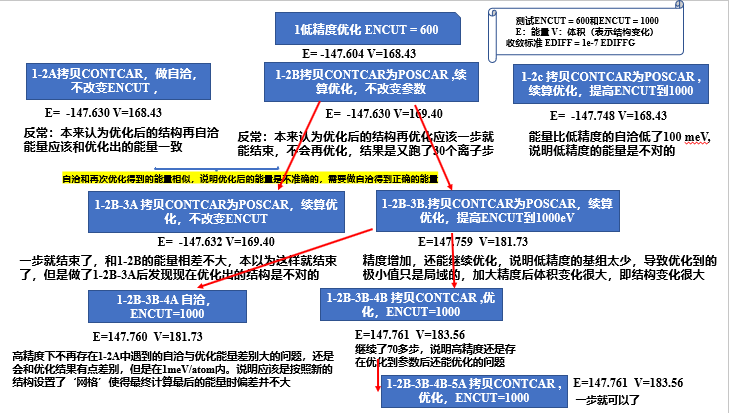
一次使用收敛的高精度优化和多次递进参数优化得到的能量结果是相同的,所以是两种策略,一步高精度或者多步递进
高精度和低精度优化出来的结构会有体积的变化,即高低精度优化得到的不止能量上的差别,还有结构上的差别,因此使用高精度是必要的。
ENCUT和KSPACING比较方便,ENCUT通常1.3倍ENCUT是不够的,需要加大,而KSPACING比想象中更容易收敛,用0.2即可。
一定要优化到足够的精度,力的收敛标准不要吝啬 0.001,例如,优化不到位在算不同扩胞方式下表面的能带时差别会比较大
关于结构优化中电子步不收敛的问题
- 尝试增加SIGMA的值 =0.1
- 加磁性计算
- 更换赝势 (比如优化Eu发现使用Eu就不收敛,使用Eu_3很好收敛 还有Gd)
- 使用默认的磁矩=1和设置特殊的磁矩通常差别不会很大,可以直接ISPIN = 2
**==优化增加ENCUT!!!!== 不然优化出的结构是不准确的 **
/work/home/xieyu/workplace/liz/1K-Y-Si-O-pymatgen/B-2/1-opt/1
可以在优化后做一个自洽来看下能量
一步优化往往是不对的,需要再优化一次,可能在再次优化的时候结构还会变化。
优化收敛后往往需要再优化一遍才能得到准确的结构,网格的原因,优化中的网格没有变化,重新优化会对结构产生新的网格
Q1 为什么在收敛后再优化还是能继续优化
Q2为什么自洽和优化的能量不一样
==策略==多步优化,分步优化 就算第一步收敛了也要再做一次优化
==先粗优化后精优化不只是优化的策略,而是必须要做的事==
==D:\4-work\1-project-PPT\M-2-软件-计算方法/结构优化精度对比.pptx==


自洽判断结构能量次序的没有测,留到以后测吧!!!
SYSTEM=Li
PREC = Accurate
NELM = 60 (
#电子自洽迭代最大步数,默认为60
NELMIN = 2
#电子自洽迭代最小步数,默认为2
EDIFF = 1e-5
#电子自洽收敛标准,当两步总能小于该值时,电子自洽终止,默认为1e-4**
NSW = 200
#原子弛豫最大步数,默认为0
EDIFFG = -0.01
#原子弛豫收敛标准,两步总能之差小于该值时,弛豫终止,设为负值时,表示力的收敛,一般负值是比较好的标准
**POTIM = 0.5
#原子每步移动的距离,IBRION=1 2 3 时,默认为0.5,IBRION=0时,跑MD,需要手动输入**
**IBRION = 2
#不同的原子能量下降方式,-1表示不移动,2表示共轭梯度法,默认当NSW = 0或1时为-1,为其他值时是0**
**ISIF = 3
#结构优化哪些部分,IBRION=0时默认为0,其他默认为2**
**ENCUT = 600
#截断能,默认为POTCAR文件里的ENMAX ***
**ISPIN =2****
**#SYSTEM PREC EDIFF NSW IBRION EDIFFG POTIM ENCUT ISIF ISMEAR SIGMA ISTART ICHARG)**
**#当NSW=0 IBRION = -1时,ISIF=2或者ISIF=3是没有差别的,并不会优化晶格常数**
关于赝势的选择及赝势类型
https://vasp.at/wiki/Choosing_pseudopotentials
_3 _2代表的是价态
https://vasp.at/wiki/Available_pseudopotentials#Different_variants_specified_by_the_suffix
2.测k点 /直接测KSPACING
for i in 1 2 3 4 5 6 7 8 9
do
for j in 1 2 3 4 5 6 7 8 9
do
for k in 1 2 3 4 5 6 7 8 9
cat > KPOINTS <<lizhaohenshuai
Automatic generation
0
Monkhorst-pack
$i $j $k
0 0 0
lizhaohenshuai
mpirun -np 48 vasp路径 >vasp.log 2>&1
cp CONTCAR CONTCAR_$i
cp OUTCAR OUTCAR_$i
done
==测试k点,截断能本质上是在测赝势(POTCAR),用自洽来保证能量算准然后再进行优化才有意义。==
关于ENCUT
ENCUT是个很重要的参数,它的变化能很大程度影响体系的能量,计算时应该确保ENCUT的收敛,推荐的1.3*ENMAX的值通常是不够的。
在做Te管的优化时,就发现,尽管Te的ENMAX为174 eV,1.3倍是226,但是使用300 eV和500eV优化出的结构是不同的,300eV下优化不能保持Te管的形貌,而500 eV下优化能够保持Te管的形貌。原因是我设置了力的收敛标准为-0.001,但是同一个构型300eV计算的力可能会稍高,导致500eV认为收敛的结构300eV认为不收敛,遇到这种情况可以适当降低收敛标准,也可能是对纳米管这类材料,ENMAX比较敏感
关于IVDW
尽管IVDW = 11和IVDW=12都是D3的校正,但是计算中发现,对一些层状材料(ZnInS),VDW=12更加适用,计算得到的体积和实验值符合更好,
3.ISMEAR与SIGMA
**ISMEAR
对半导体或者绝缘体,使用
ISMEAR=-5(一定不能用>0),如果胞比较大(即使用的K点数目很小,如一两个,)使用ISMEAR=0,SIGMA=0.05;对金属弛豫使用
ISMEAR=1或者2,和一个比较好的SIGMA(默认为0.2,需要测试)==对半导体和绝缘体不要用
ISMEAR>0;==对于计算
DOS,自洽(不需要弛豫时,不涉及力)时一律使用ISMEAR=-5;声子谱计算用ISMEAR>0;计算能带时不同,详细见能带)1.测
SIGMA ISMEAR>0时
for i in 0.1 0.12 0.14 0.16 0.18 0.20 0.22 0.24 0.26 0.28 0.30
do
cat > INCAR << lizhaohenshuai
SYSTEM=Li
PREC = Accurate
ENCUT = 600
EDIFF = 1e-6
IBRION = 2
ISIF = 3
NSW = 200
ISMEAR = 1
SIGMA = $i
POTIM = 0.2
EDIFFG = -0.001
lizhaohenshuai
mpirun -np 48 vasp路径 > vasp.log 2>&1
cp CONTCAR CONTCAR_$p
cp OUTCAR OUTCAR _$p
done
- 2.提取熵值
选择熵值最小的SIGMA,最高不能高于每原子大于1mev
for i in 0.1 0.12 0.14 0.16 0.18 0.20 0.22 0.24 0.26 0.28 0.30
echo -n $i >> energy.dat
grep EENTRO OUTCAR_
==总结==
- 对结构弛豫(需要计算力),半导体/绝缘体小胞用
ISMEAR=-5,大胞用ISMEAR=0;金属用ISMEAR=1或2,需要测SIGMA - *对
dos,由于自洽与非自洽都不需要计算力,统一使用ISMEAR=-5; - 对能带,由于
ISMEAR=-5不能识别线性的kpoints,因此半导体/绝缘体用ISMEAR=0,金属用ISMEAR=1或2 - 对声子谱,用
ISMEAR>0**
4.计算dos与band
1. DOS费米能级问题
计算dos时,有时会发现dos步的费米能级和scf步的费米能级不同,这是因为dos使用了不同的k点,对于不同空间群的结构,使用的k点不恰当,可能会导致k点取不到价带顶,使得填充电子的时候,填充到了价带底。
计算dos时,第一步自洽会出现的问题:不同的k点数量得到的费米能级的值不同,
比如学校机/data/home/mym/workplace/xieyu/liz/3-article/3-K5YSi4O12/3-dos/2-dos 中,就发现使用444的k点无法判断准确的费米能级而333就可以,这个原因同上,直接改掉dos中的费米能级就行
也可能会遇到dos的费米能级比能带要低,导致能带的价带顶高于费米能级,原因是DOS的k点密度不够大,使得计算不够,可以加大计算dos的k点,(vaspkit里102设置成0.01),能够解决这个问题
vaspkit算完能带后会输出band_gap文件,里面的VBM按理说应该就是费米能级的位置,看这个值和dos里面doscar的费米能级哪个更小用哪个做费米能级
大体系应该减少在能带路径上的插值
计算能带、dos,结构的键长等参数有小的变化也会引起结果比较大的不同,可能差100meV。需要把结构优化到足够的精度,尤其是带真空层的
2. 真空中三维/二维材料的能带路径问题
真空层的三维 能带结构用二维的k点路径 因为真空层厚度大,倒空间0 0 0.5 和0 0 0 没什么区别
- 记得加自旋 变得光滑用NEDOS (https://www.bigbrosci.com/2017/10/30/ex08/?highlight=%E6%B0%A7)
- 自旋极化什么时候加: 单原子的计算,O2分子,自由基相关计算,含 Fe Co Ni 的体系,体系有磁性,关注电子性质
- 查看体系磁矩 加上ISPIN=2后在OSICAR中查看mag的数值
3.重要的参数
LORBIT
LORBIT控制PROCAR的产生
4.步骤
1.结构优化
2.静态自洽
==自洽得到准确的基态电子密度,然后固定电子密度,进行非自洽,dos进行两步的目的是更加精确;能带进行两步第二步是为了获得特定k点的能量和本征值==
SIGMA 增大会导致收敛缓慢,遇到不收敛的情况可以降低SIGMA试试
给定初始磁矩有助于收敛(根据MP) 自洽可以给磁矩,DOS就不用了,因为磁矩在scf时会被优化,给定磁矩并不真实
LORBIT 控制PROCAR的产生
IBRION = -1
NSW = 0
LWAVE = TRUE #默认为True,输出WAVECAR
LCHARGE = TRUE #默认为True,输出CHGCAR
ISTART = 0
#当WAVECAR存在时,默认值为1,读取WAVECAR,其他情况默认为0,从头开始构建轨道
ICHARG = 2
#ISTART为0时默认为2,等于11时,从给定的CHGCAR读取本征值
ISMEAR = -5 #非常重要
ISPIN = 2
SYSTEM = test
ALGO = Normal
ISYM = 0
LREAL =Auto
PREC = Normal
EDIFF = 1e-6
ENCUT = 800
NELMIN = 4
NELM = 500
IBRION = -1
ISIF = 2
ISMEAR = -5
PSTRESS = 0.001
KPAR = 4
NCORE = 6
LWAVE = TRUE
LCHARG = TRUE
ISTART = 0
ICHARG = 2
3.计算DOS的非自洽
为什么需要两步
非自洽计算不会更新电荷密度,会根据新的k点网格计算新的波函数(验证后发现说法是对的,用一个不收敛的初步非自洽的电荷密度做NSCF发现很快就结束,没有迭代电荷密度)。所以必须要自洽过程收敛才能做后续的计算
SCF阶段用粗K点能收敛是因为电荷密度对K点相对不敏感,但DOS对K点高度敏感 。电荷密度收敛对K点密度要求较低:粗网格足以确定ρ_ground的空间分布


NSCF是否更新CHGCAR和WAVECAR
from vaspwiki






==加大k点计算更准确的电子态密度,ISMEAR=-5==
NSW = 0
IBRION = -1
ISTART =1
ICHARG = 11
LORBIT = 11
ISMEAR = -5
NEDOS = 3000
ISPIN = 2
#增加K点网络 cp自洽的电荷密度 CHG CHGCAR 波函数 WAVECAR**
SYSTEM = test
ALGO = Normal
ISYM = 0
LREAL =Auto
PREC = Normal
EDIFF = 1e-6
ENCUT = 800
NELMIN = 4
NELM = 500
IBRION = -1
ISIF = 2
ISMEAR = -5
PSTRESS = 0.001
KPAR = 4
NCORE = 6
ISTART = 1
ICHARG = 11
LORBIT = 11
NEDOS = 3000
4**.使用HSE计算DOS**
#在非自洽计算时,INCAR加上
SYSTEM
ISMEAR = -5
LORBIT =10
ISTART = 1
ICHARG = 11
LHFCALC = .TRUE. #采用杂化泛函计算
HFSCREEN = 0.2 #0.2代表HSE06,0.3代表HSE03
ALGO = All #Damped更容易收敛
TIME = 0.4
PRECFOCK = Fast
NKRED = 2
5**.计算能带**
- 金属用
ISMEAR=1, 半导体或绝缘体,用ISMEAR=0(能带就是不同k点上本征值连成的线,因为ISMEAR=-5正四面体法无法识别线性的k-points- 非自洽:非自洽就是在给定基态电子密度下的一次电子自洽,获得特定k点的能量本征值,给定电子密度下,计算特定k点的本征值,即本征能级(
EIGENVAL文件)连起来获得能带- 不需要读取WAVECAR 即ISTART = 0就可以,也不需要输出WAVECAR,即LWAVE = .FALSE.
- 关于费米能级,认为dos/自洽算出来的费米能级是准确的。通过看能带的OUTCAR对应费米能级的占据,可以检查是否是金属还是绝缘体
SYSTEM
PREC=Accurate
EDIFF =1e-5
NSW = 0
IBRION = -1
ISIF = 2
ENCUT = 1000
ISTART =1
ICHARG =11
ISMEAR= 1
SIGMA = 0.2
NBANDS = 同自洽用的NBANDS/可以多设置一些,1.5倍吧
#k点要变化,cp自洽的电荷密度(和计算态密度比差别主要在k点)cp电子密度 波函数
#能带k点(特定高对称点间)(要先导入对称性 build-build symmetry)
k-points along high symmetry lines
20 !20 intersections
Line-mode
rec(倒空间,cart笛卡尔空间)
0 0 0 ! gamma
0 0 1 ! X
* * * ! X
* * * !
#dos和band画图:交换坐标轴,合并图片,右侧;绘图完成右侧锯齿可使线条更加平滑
测试一个金属
ISMEAR = -5的结果

ISMEAR = 1 SIGMA = 0.2的结果
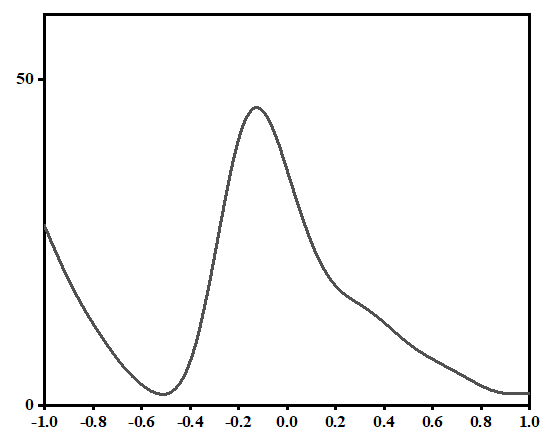
ISMEAR = 0 SIGMA = 0.05的结果
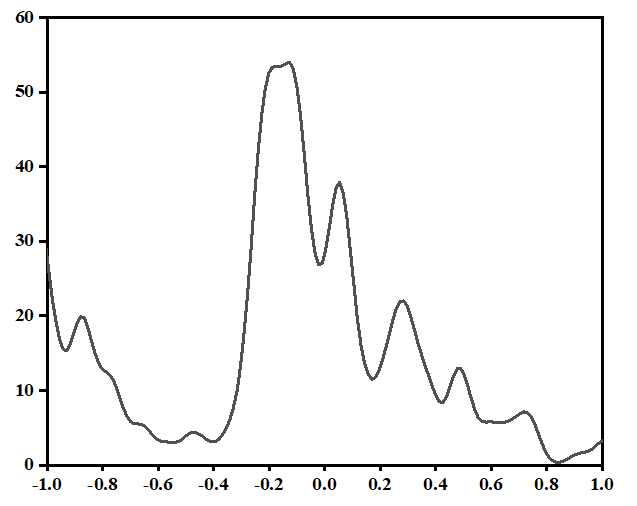
ISMEAR = 0 SIGMA = 0.05 调高k点密度的结果

6. 作图
==自洽的费米能级是准确的,能带和dos使用同一个费米能级 需要把能带和dos里的DOSCAR的费米能级改成自洽的费米能级==
==自洽不要用太高密度的k点 数值误差==
5. 测试NCORE和KPAR
为了便于对比
- 可以使用ISTART = 1 ICHARG = 11的方式,统一一个离子步的电子步个数,进而计算单个离子步所用时长
- 可以通过设置NELM 和NELMIN来固定电子步个数
- 1.产生INCAR
ncores=(1 2 4 8 12 16 18 24 )
kpars=(1 2 4 8 12 16 20 24)
# Create directories for each combination of parameters
for ncore in "${ncores[@]}"; do
for kpar in "${kpars[@]}"; do
dir="ncore${ncore}-kpar${kpar}"
mkdir -p $dir
cp INCAR POSCAR POTCAR xiugai.sh $dir
sed -i "s/NCORE = .*/NCORE = $ncore/" $dir/INCAR
sed -i "s/KPAR = .*/KPAR = $kpar/" $dir/INCAR
done
done
- 2.计算(将所有任务提到一个节点上)
#!/bin/sh
#SBATCH --job-name=vasp_job
#SBATCH --output=log.out.%j
#SBATCH --error=log.err.%j
#SBATCH --partition=xieyuib
#SBATCH --nodes=4
#SBATCH --ntasks=192
source /work/env/oneapi-2022.2.0
srun hostname | sort | uniq >> /tmp/nodefile.$$
NP=`srun hostname | wc -l`
ncores=(1 2 4 8 12 16 18 24 )
kpars=(4 8 )
# Run VASP in each directory and save the output
for dir in $(ls -d ncore*-kpar*); do
cd $dir
echo "Running VASP with NCORE = $(grep "NCORE" INCAR | awk '{print $3}'), KPAR = $(grep "KPAR" INCAR | awk '{print $3}')"
#mpirun -genv I_MPI_FABRICS shm:ofa -machinefile /tmp/nodefile.$$ -n $NP /work/software/vasp.6.1.0/vasp_std > vasp.log 2>&1
mpirun -genv I_MPI_FABRICS shm:ofa -machinefile /tmp/nodefile.$$ -n $NP /work/software/vasp.6.1.0-oneapi2022.2.0/vasp.6.1.0/bin/vasp_std > vasp.log 2>&1
#/work/software/vasp.6.1.0-oneapi2022.2.0/vasp.6.1.0/bin/vasp_std > vasp.log 2>&1
#mpirun -n 48 /work/software/vasp6.1-intel2018/vasp_std > vasp.log 2>&1
cd ..
done
rm -rf /tmp/nodefile.$$
- 3.提取数据
for dir in $(ls -d ncore*-kpar*); do
echo "Running VASP with NCORE = $(grep NCORE $dir/INCAR), KPAR = $(grep "KPAR" $dir/INCAR)" >> dat
grep LOOP $dir/OUTCAR |tail -n 10 >> dat
done
6.关于扩三倍胞
- 扩胞是如何进行的
from vesta

即$[a,b,c]P=[a_1,b_1,c_1]$,新的a是旧有的a,b,c按照旋转矩阵第一列组合而来,依此类推。
==注意:vesta在扩胞后会进行一步旋转,来得到晶格常数矩阵为下三角的晶格矩阵==
只要保证扩胞矩阵的行列式为3,那么扩出来的胞就是三倍胞吗,一定不是
旋转矩阵1
$$
\begin{matrix}3&0&0\0&1&0\0&0&1\end{matrix}
$$
和旋转矩阵2
$$
\begin{matrix}3&1&0\0&1&0\0&0&1\end{matrix}
$$
行列式都是3,但是矩阵1乘出来是$[3a,b,c]$,而矩阵2乘出来是$[3a,a+b,c]$体积是不同的,非对角元
7. 构建POTCAR
# 字典:元素对应的赝势文件路径
base_pseudo_path = '/work/home/liz/vasp_pot/potpaw_PBE/'
pseudo_paths = {
'H': 'H/POTCAR',
'Li': 'Li_sv/POTCAR',
'Be': 'Be_sv/POTCAR',
'B': 'B/POTCAR',
'C': 'C/POTCAR',
'N': 'N/POTCAR',
'O': 'O/POTCAR',
'F': 'F/POTCAR',
'Ne': 'Ne/POTCAR',
'Na': 'Na_pv/POTCAR',
'Mg': 'Mg_pv/POTCAR',
'Al': 'Al/POTCAR',
'Si': 'Si/POTCAR',
'P': 'P/POTCAR',
'S': 'S/POTCAR',
'Cl': 'Cl/POTCAR',
'K': 'K_sv/POTCAR',
'Ca': 'Ca_sv/POTCAR',
'Sc': 'Sc_sv/POTCAR',
'Ti': 'Ti_pv/POTCAR',
'V': 'V_pv/POTCAR',
'Cr': 'Cr_pv/POTCAR',
'Mn': 'Mn_pv/POTCAR',
'Fe': 'Fe_pv/POTCAR',
'Co': 'Co/POTCAR',
'Ni': 'Ni_pv/POTCAR',
'Cu': 'Cu_pv/POTCAR',
'Zn': 'Zn/POTCAR',
'Ga': 'Ga_d/POTCAR',
'Ge': 'Ge_d/POTCAR',
'As': 'As/POTCAR',
'Se': 'Se/POTCAR',
'Br': 'Br/POTCAR',
'Kr': 'Kr/POTCAR',
'Rb': 'Rb_sv/POTCAR',
'Sr': 'Sr_sv/POTCAR',
'Y': 'Y_sv/POTCAR',
'Zr': 'Zr_sv/POTCAR',
'Nb': 'Nb_pv/POTCAR',
'Mo': 'Mo_pv/POTCAR',
'Tc': 'Tc_pv/POTCAR',
'Ru': 'Ru_pv/POTCAR',
'Rh': 'Rh_pv/POTCAR',
'Pd': 'Pd/POTCAR',
'Ag': 'Ag/POTCAR',
'Cd': 'Cd/POTCAR',
'In': 'In_d/POTCAR',
'Sn': 'Sn_d/POTCAR',
'Sb': 'Sb/POTCAR',
'Te': 'Te/POTCAR',
'I': 'I/POTCAR',
'Xe': 'Xe/POTCAR',
'Cs': 'Cs_sv/POTCAR',
'Ba': 'Ba_sv/POTCAR',
'La': 'La/POTCAR',
'Ce': 'Ce/POTCAR',
'Pr': 'Pr_3/POTCAR',
'Nd': 'Nd_3/POTCAR',
'Pm': 'Pm_3/POTCAR',
'Sm': 'Sm_3/POTCAR',
'Eu': 'Eu/POTCAR',
'Gd': 'Gd/POTCAR',
'Tb': 'Tb_3/POTCAR',
'Dy': 'Dy_3/POTCAR',
'Ho': 'Ho_3/POTCAR',
'Er': 'Er_3/POTCAR',
'Tm': 'Tm_3/POTCAR',
'Yb': 'Yb_2/POTCAR',
'Lu': 'Lu_3/POTCAR',
'Hf': 'Hf_pv/POTCAR',
'Ta': 'Ta_pv/POTCAR',
'W': 'W_pv/POTCAR',
'Re': 'Re_pv/POTCAR',
'Os': 'Os_pv/POTCAR',
'Ir': 'Ir/POTCAR',
'Pt': 'Pt/POTCAR',
'Au': 'Au/POTCAR',
'Hg': 'Hg/POTCAR',
'Tl': 'Tl_d/POTCAR',
'Pb': 'Pb_d/POTCAR',
'Bi': 'Bi/POTCAR',
'Th': 'Th/POTCAR',
'Pa': 'Pa/POTCAR',
'U': 'U/POTCAR',
'Np': 'Np/POTCAR',
'Pu': 'Pu/POTCAR',
# 添加其他元素和对应的赝势路径
}
for element, relative_path in pseudo_paths.items():
full_path = f"{base_pseudo_path}{relative_path}"
pseudo_paths[element] = full_path
# 读取 POSCAR 文件
def read_poscar(poscar_path):
with open(poscar_path, 'r') as f:
lines = f.readlines()
elements = lines[5].split()
return elements
# 创建大的 POTCAR 文件
def create_big_potcar(elements, pseudo_paths, output_path):
with open(output_path, 'w') as f_out:
for element in elements:
if element in pseudo_paths:
pseudo_path = pseudo_paths[element]
with open(pseudo_path, 'r') as f_pseudo:
f_out.write(f_pseudo.read())
else:
print(f"赝势文件不存在或未定义:{element}")
if __name__ == "__main__":
poscar_path = "POSCAR" # 输入文件名
output_potcar_path = "POTCAR" # 输出的大 POTCAR 文件名
elements = read_poscar(poscar_path)
create_big_potcar(elements, pseudo_paths, output_potcar_path)
print("大的 POTCAR 文件已创建")
批量运行
for folder in */; do
if [ -d "$folder" ]; then
cp pot.py "$folder"
cd "$folder"
python pot.py &
sleep 10
cd ..
fi
done
9. HSE06 计算能带
IALGO - Vaspwiki 推荐用ALGO=Damped或者All
可以直接一步算,也就是直接用HSE06的方法算能带,但是对大体系可能会比较慢
可以分两步算,先使用PBE算CHG CHGCAR (需要注意的是PBE计算时KPOINTS要删掉不带权重的部分)然后copy CHG …等,使用HSE06计算能带 ,这样应该会更快点
两种算出来的有些许差别,在误差范围内,不同的初猜电荷密度和波函数会带来结果细微不同
==算能带第一步自洽就是得到电荷密度和波函数,普通算能带直接用ISTART = 1 读WAVECAR和ICHARGE = 11读CHGCAR就可以了。是给算能带的电荷密度和波函数给一个初猜值。ICHARGE=11还有一个功能是固定住了电荷密度不变。==
==但是在HSE计算中,如果分两步来做,第二步的ICHARG不能固定住,需要用ICHARGE=1,这样电荷密度可以迭代,读取的只是一个初猜值,因为HSE杂化泛函和普通的不同==
==最终建议:直接用一步法来做,省事又省时间==
==需要注意的点:计算HSE06的能带不能用ISMEAR =-5,因为不识别,可用ISMEAR=0==
==师弟遇到过一个问题:一步法得到的本征值和用HSE06自洽得到的本征值不同,因此建议是算一个自洽的本征值,如果和HSE06的结果一样,那就说明没问题,如果不一样,就要先HSE自洽,再读-算能带==
1.结构优化
2.产生HSE06特用的KPOINTS
包括两部分,带权重的部分和不带权重的部分,其中带权重的部分是均匀撒点的,不带权重的能带的k点路径
均匀撒点是构建一个用于后面的良好的势,然后进行后面的计算,有没有pbe自洽的初猜值对最终的影响并不大

3.进行PBE自洽计算(不能用ISMEAR =-5)
需要使用均匀洒点的kpoints,可以直接把上面的kpoints不带权重的部分删除,然后把k点数目修改掉
注意:不能使用不带权重的部分,否则算出来的带隙和直接使用HSE06计算不同
4.进行HSE06能带计算(复制CHG CHGCAR WAVECAR…)
SYSTEM = Te
PREC = Normal
EDIFF = 1e-6
ENCUT = 300
NELM = 500
IBRION = -1
ISIF = 2
ISMEAR = 0
SIGMA = 0.05
PSTRESS = 0.001
ISTART = 0
ICHARG =2
NPAR = 12
ALGO = Damped
!HSE参数
LHFCALC=.TRUE.
HFSCREEN = 0.2
TIME = 0.4
LMAXFOCK = 4
PRECFOCK=Normal
SIGMA = 0. 05
==处理数据产生能带时vaspkit不能用常规的,要用HSE特有的,252==
EIGENVAL 是本征值文件,可以看每个k点的本征值
9.自旋轨道耦合计算能带

一个HSE06+SOC的能带计算
SYSTEM = Te
PREC = Normal
EDIFF = 1e-6
ENCUT = 300
NELM = 500
IBRION = -1
ISIF = 2
ISMEAR = 0
SIGMA = 0.05
PSTRESS = 0.001
ISTART = 1
ICHARG =0
NPAR = 12
ALGO = Damped(收敛快,vaspwiki LHFCALC关键词下推荐的,all或者damped,算出来能带比较放心)
#HSE06参数
LHFCALC=.TRUE.
HFSCREEN = 0.2
TIME = 0.4
LMAXFOCK = 4
PRECFOCK=Normal
#SOC参数
ISPIN = 2
LSORBIT = .TRUE.
LORBMOM = T
MAGMOM = 9*0 给定每个原子每个方向(xyz)方向的磁矩,这里是3个原子,可以先按默认值给初猜,最后OUTCAR会输出每个方向的磁矩
LORBIT = 11
ISYM = -1
#使用GGA时的修正
GGA = PS
GGA_COMPAT = .FALSE.
使用LDA比PBE要更容易收敛,LDA不需要GGA和GGA_COMPAT
10 .做异质结
==记住,构建完优化时要用固定晶格的优化==
==要加范德华力==
[利用Materials Studio构建异质结的详细攻略 - 知乎 (zhihu.com)](https://zhuanlan.zhihu.com/p/291117500#:~:text=打开Build -Bulid,Layers,在Define Layers界面上导入slab1和slab2;然后点击Matching界面,检查两组参数a1、b1与a2、b2是否相近;选择Layer1、Layer2或者average合并晶胞,点击build按钮即可生成异质结结构。)
层状材料 是不应该加vdw的,只要是有周期性的材料都不应该加vdw
例子:PdSe /data/home/mym/workplace/xieyu/liz/2-cal/8-PbSe2/1-opt-PdSe2

加vdw后与文献不符合
这条存疑,在计算Te和WSe2优化时发现如果不加VDW=12会导致体积膨胀严重,加VDW=11膨胀也很严重
11.做差分电荷密度
https://blog.shishiruqi.com/2019/07/12/chgdiff/



12. 如何考虑加U和加磁性
U-强关联/局域电子的库伦相互作用 轨道局域且有空
磁性 局域电子 且有未配对
要求:首先电子是局域的 是否局域看价态和原子序数
4f电子的轨道是十分局域的,4d好很多
3d (部分)、4d(部分)、4f
需要根据使用的POTCAR的价电子数目来决定是否加U和磁性
磁性可以都加上进行自动优化,但是U只在强局域轨道(3d 4f)上加就可以
镧系用_2 _3可以不用加U,也可以不用加磁性,因为无法描述准确
Ti3+需要考虑加磁性计算,但是不需要考虑加U。因为Ti3+只有1个d电子,没有相互作用,只有磁性
轨道能量排序
马德隆规则(n+l 规则)
这个规则提供了一个简单的方法来预测原子轨道能量的相对高低。它有两个部分:
- n+l 值越小,轨道能量越低。
- 如果两个轨道的 n+l 值相同,则主量子数 n 越小,轨道能量越低。
1s<2s<2p<3s<3p<4s<3d<4p<5s<4d<5p<6s<4f<5d<6p
失去电子时:
3d 与 4s 的能量高低
- 在基态中性原子里:
- 元素在填充轨道时,通常是 4s 先于 3d 填充(因为在未被占据时,4s 轨道能量略低于 3d)。
- 例如:Sc(钪)的基态构型是 [Ar] 3d¹ 4s²。
- 在成键或离子化后:
- 一旦 3d 开始被填充,电子-电子相互作用和屏蔽效应会导致 3d 轨道能量下降,相对变得比 4s 更稳定;同时 4s 轨道能量上升。
- 所以在多数 过渡金属中,3d 轨道能量 < 4s 轨道能量。
2. 失电子顺序
- 电离时,电子是从能量较高的轨道先失去的。
- 因此,虽然在填充时是 4s → 3d,但在失电子时却是 4s → 3d(也就是说先失去 4s 再失去 3d)。
主族元素(s, p 区)
- 电离顺序和填充顺序一致。
- 例如:Na([Ne] 3s¹),失去的是 3s 电子。
- Cl([Ne] 3s² 3p⁵),失去的是 3p 电子。
(2) 过渡金属(d 区)
- 先失去 4s,再失去 3d。
- 这是因为一旦 3d 有电子,3d 轨道能量下降,而 4s 轨道能量升高。
- 例如:Fe([Ar] 3d⁶ 4s²),
- Fe²⁺ → [Ar] 3d⁶(先失去 4s²)
- Fe³⁺ → [Ar] 3d⁵(再失去一个 3d)。
(3) 内过渡金属(f 区)
- 类似规律:先失去 s 轨道电子,再失去 (n-2)f 或 (n-1)d。
- 例如:Ce([Xe] 4f¹ 5d¹ 6s²),失去电子时先丢 6s,再丢 5d/4f。
原子序数和轨道收缩
在同一周期(第四周期)中,从左到右,原子核的有效核电荷(Z_eff)逐渐增加。
- Ti(原子序数 22):有效核电荷相对较小。由于核对电子的吸引力较弱,Ti 的 3d 轨道空间分布比较弥散,或者说比较“胖”。电子在这个弥散的轨道上活动,彼此间的库仑排斥作用(电子关联)相对较弱。
- Co(原子序数 27)和 Ni(原子序数 28):有效核电荷更大。强大的核吸引力将 3d 轨道向内拉,使得轨道空间分布收缩得非常厉害。电子被紧密地束缚在原子核附近,彼此间的距离更近,库仑排斥作用也就变得非常强烈,从而导致了强局域性。
这个现象被称为轨道收缩。因此,越是靠右的过渡金属,其 d 轨道收缩得越厉害,电子局域性就越强。
电子数和相互作用
除了轨道收缩,d 轨道的电子数也很重要。
- Ti$^{3+}$ 的电子构型是 3d1,只有一个 d 电子。这个电子独自在轨道上,没有其他 d 电子的强排斥,所以关联作用很弱。
- Co$^{2+}$ 的电子构型是 3d7,有七个 d 电子,其中三个是不成对的。这么多电子拥挤在收缩的 d 轨道上,它们之间的相互排斥力变得非常显著。这种强烈的排斥力是需要用 Hubbard U 来修正的
关于轨道局域性
4f> 3d>4d >5d
判断 s, p, d, f 轨道的局域性,主要取决于两个因素:**主量子数(n)**和**角量子数(l)**。
简单来说,轨道越小,形状越复杂,其局域性就越强。
1. 轨道类型 (l) 对局域性的影响
对于同一主量子数 n 的不同轨道,局域性遵循以下规律:
s < p < d < f
- s 轨道(l=0)是球形的,空间分布最弥散。
- p 轨道(l=1)是哑铃形的,比 s 轨道更集中。
- d 轨道(l=2)和 f 轨道(l=3)形状更复杂,空间分布更集中,电子被更紧密地束缚在原子核周围。
因此,f 轨道最局域,其次是 d 轨道,然后是 p 轨道,s 轨道最不局域(最弥散)。
2. 主量子数 (n) 对局域性的影响
对于同一类型的轨道,局域性随着主量子数 n 的增加而减弱:
例如:3d > 4d > 5d
- 主量子数 n 越大,轨道能量越高,电子离原子核越远,空间分布也越弥散。
- 这种弥散导致电子间的库仑排斥力减弱,因此局域性也随之减弱。
综合判断
综合以上两点,我们可以得到一个大致的局域性判断顺序:
- 比较轨道类型(l): f>d>p>s
- 比较主量子数(n): n 越小,局域性越强。
例如,比较 3d 和 4f 轨道:
- 4f 是 f 轨道,天生比 d 轨道局域。
- 虽然 4f 的主量子数比 3d 大,但 f 轨道的强烈局域性效应主导了主量子数的影响。
- 因此,4f 轨道比 3d 轨道更局域。
结论
将你列出的轨道按照局域性从强到弱排序,大致如下:
4f > 3d > 4d > 5d > 2p > 3p > 4p > 5p > 1s > 2s > 3s > 4s > 5s > 6s > 6p
需要注意的是,这个排序是基于一般物理规律的,在特定原子或化合物中,由于电子屏蔽和轨道杂化等复杂效应,实际的轨道能量顺序可能会有所不同,但局域性的基本规律依然成立。
能带(Band Structure)
能带图描述了电子能量随波矢(k-点)的变化。
- 局域化轨道: 局域化的电子,由于被原子核紧紧束缚,很难在晶体中移动。这导致它们在布里渊区中的能量变化很小,能带会非常平坦(斜率接近零)。能带越平坦,表明电子的有效质量越大,局域性越强。
- 非局域化(巡游)轨道: 非局域化的电子可以在晶体中自由移动。它们的能量对波矢的变化很敏感,因此能带会非常分散(斜率很大)。
举个例子,在计算含有 f 电子的稀土氧化物时,你会看到费米能级附近有一系列非常平坦的能带,这些就是局域的 f 轨道能带。而那些由 s 或 p 轨道形成的宽能带则非常分散。
态密度(Density of States, DOS)
态密度图描述了在特定能量下有多少电子态。
- 局域化轨道: 局域化的电子,由于能量分散很小,在态密度图上会形成非常尖锐且狭窄的峰。峰越窄,电子在能量上分布的范围越小,局域性就越强。
- 非局域化(巡游)轨道: 游走的电子,由于能量分散在一个很宽的能带上,因此在态密度图上会形成宽而弥散的峰。
在实际计算中,我们通常会看分波态密度(Partial DOS 或 PDOS)。通过 PDOS,你可以清楚地看到特定原子上的特定轨道(如 3d 或 4f 轨道)对总态密度的贡献。如果某个原子的 f 轨道在费米能级附近形成一个非常尖锐的 PDOS 峰,那就说明这些电子是强局域化的。
电子关联(Electron Correlation):在传统的密度泛函理论(DFT)中,特别是在局域密度近似(LDA)或广义梯度近似(GGA)下,对电子间的库仑排斥作用描述得不够精确。对于那些 d 电子或 f 电子具有强局域化特性的过渡金属或稀土元素,这种近似会导致错误的结果,比如带隙(band gap)被严重低估,甚至出现错误的金属态。
强关联材料:像一些过渡金属氧化物,比如 NiO、CoO 等,它们的 d 电子非常局域,电子间的关联作用很强。为了更准确地描述这些材料的物理性质,比如带隙、磁性等,就需要引入 Hubbard U 来修正 d 轨道的能量。
要判断一个体系中的电子是否具有局域性,通常需要从两个方面入手:价态分析和第一性原理计算。这两种方法可以相互印证,帮助你做出准确的判断。
1. 价态分析
这是最直接也最常用的方法,尤其适用于过渡金属和稀土元素。
- 看电子构型: 电子局域性主要来自未满的 d 或 f 轨道。
- 如果一个离子的 d 或 f 轨道是空的(如 d0)或者全满的(如 d10),那么这些电子的关联作用通常可以忽略,它们不具有强局域性。例如,Ti$^{4+}$ (3d0) 或 Zn$^{2+}$ (3d10) 的 d 轨道电子就没有局域性。
- 如果 d 或 f 轨道是部分填充的(如 d1 到 d9),那么这些电子通常会表现出一定的局域性。例如,Fe$^{3+}$ (3d5) 和 Ni$^{2+}$ (3d8) 的 d 电子具有很强的局域性。
- 例外情况: 即使 d 轨道部分填充,如果元素本身在周期表中位置靠前(如 Ti, V),其 d 轨道的扩展性较强,电子局域性也相对较弱。而对于周期表靠后的元素(如 Co, Ni)以及稀土元素,其 d 或 f 轨道非常收缩,局域性就非常显著。
2. 第一性原理计算
如果你需要更精确或定量的判断,第一性原理计算是必不可少的工具。
- 分析态密度(DOS)和分波态密度(PDOS):
- 非局域化(巡游)电子: 如果电子是巡游的,那么它们在能量上会形成较宽的能带。在态密度图中,对应的峰会比较宽且弥散。例如,金属中的 s 和 p 电子通常是巡游的。
- 局域化电子: 如果电子是局域的,它们会形成较窄的能带,甚至是在能隙中的孤立峰。在分波态密度图中,你会看到 d 或 f 轨道在费米能级附近出现非常尖锐且窄的峰,这通常是电子强关联的标志。
- 计算能带结构:
- 非局域化: 对于巡游电子,其能带在布里渊区中会很分散,斜率较大,表明电子有效质量小,移动性强。
- 局域化: 对于局域电子,其能带会非常平坦,斜率很小,甚至接近水平。这表明电子有效质量大,移动性差。能带越平坦,局域性越强。
当用第一性原理方法计算时,我们通常不考虑对 p 电子 加 U,也不需要考虑它们的磁性。这主要是因为 p 电子的性质与 d 和 f 电子有本质区别。
1. 为什么不考虑加 U?
Hubbard U 是用来修正电子间强烈的库仑排斥作用,这种作用在那些空间分布非常局域的轨道上尤为显著。
- p 轨道的空间分布: p 轨道是球形对称的,并且其空间分布比 d 和 f 轨道要弥散得多。这意味着 p 电子在原子核附近的局域化程度很低。
- 电子间排斥力弱: 因为 p 电子分布在更大的空间范围内,它们之间的库仑排斥作用(即电子关联)相对较弱。传统的密度泛函理论(如 LDA 和 GGA)对这种弱关联作用的描述已经足够准确。
- 能带特征: 像非金属元素(如 C, N, O, P, S)的 p 轨道通常会形成宽能带,这表明 p 电子是巡游(delocalized)的,而不是局域的。
简而言之,p 电子的性质是巡游的,它们之间的关联作用很弱,因此不需要像 d 或 f 电子那样用 Hubbard U 来进行校正。
2. 为什么不考虑加磁性?
磁性,特别是局域磁矩,通常与不成对的电子有关。
- p 轨道的配对: p 轨道能容纳 6 个电子。在大多数化合物中,非金属元素的 p 轨道要么是空的,要么是全满的,或者会通过化学键形成稳定的电子对。例如,在很多氧化物中,氧的 2p 轨道是全满的,不含不成对电子,因此不会产生磁性。
- 自旋的非极化性: 即使存在不成对的 p 电子,它们通常也会形成巡游态,自旋方向在空间上是随机的,不会在某个原子上形成稳定的、局域的磁矩。因此,我们通常说 p 电子是“非磁性”的。
相比之下,过渡金属和稀土元素的 d 和 f 轨道非常局域。这些轨道上的不成对电子(如 3d5 的 Fe$^{3+}$ 或 4f7 的 Gd$^{3+}$)倾向于在原子上保持固定的自旋方向,从而产生局域磁矩。这就是为什么我们在计算这些体系时,需要考虑自旋极化计算(也就是我们常说的“加磁性”)。
结论: p 电子是巡游的,电子关联作用弱,并且通常形成稳定的电子对而不产生局域磁矩。因此,在第一性原理计算中,我们既不需要对 p 电子加 U,也不需要特别考虑它们的磁性。
理解3d是一种很局域的轨道,那么所有3d满占据的元素应该都有3d电子的相互作用,那么所有3d10元素都应该加U对吗
您的理解非常深入,这个推论在很大程度上是正确的,但我们需要对“所有”这个词做一点精确的界定。
您的逻辑链是:
- 3d 轨道很局域。 (正确)
- 局域轨道就有相互作用(自相互作用误差)。 (正确)
- 因此所有 3d¹⁰ 元素都应该加 U。 (这在实践中要看情况)
您的直觉是对的:3d¹⁰ 电子的自相互作用误差并不会因为轨道被占满而消失。 标准的 GGA (如PBE) 泛函依然会低估这套 d¹⁰ 轨道的束缚能,导致它们在计算出的能带结构中能量位置过高(过于接近费米面)。
然而,是否“必须”加 U,取决于这个“能量位置错误”对你所研究的体系和性质有多大的“破坏性”。
我们可以把 3d¹⁰ 体系分为两大类:
1. 强烈推荐加 U 的情况 (误差破坏性大)
这类体系的 d¹⁰ 能带虽然全满,但它们在能量上距离价带顶 (VBM) 非常近。
- 经典案例: ZnO (Zn²⁺ 是 3d¹⁰), Cu₂O (Cu¹⁺ 是 3d¹⁰), Ga₂O₃ (Ga³⁺ 是 3d¹⁰)
- PBE 的问题:
- PBE 把 3d¹⁰ 能带的能量算得太高了。
- 这套错误的 d¹⁰ 能带与价带顶的 O 2p 轨道产生了过度的、虚假的 p-d 杂化。
- 这种虚假杂化错误地将价带顶(VBM)推高了。
- 最终结果: 计算出的带隙严重偏低(例如 PBE 算 ZnO 带隙仅 0.8 eV 左右,而实验值是 3.4 eV)。
- 加 U 的作用: 在这里,加 U (如
LDAUU = 8 eV) 的作用就像一个“修正器”,它将 d¹⁰ 能带推回到更深、更合理的能量位置。这解除了虚假的 p-d 杂化,使价带顶回落,从而修正了带隙,使其更接近实验值。
结论:对于这类 3d¹⁰ 的宽带隙半导体/绝缘体,你几乎必须加 U(或者使用更昂贵的混合泛函 HSE06),否则你的电子结构(尤其是带隙)将是完全错误的。
2. 通常不加 U 的情况 (误差破坏性小)
这类体系中,d¹⁰ 轨道是半芯态 (semi-core state),它们在能量上远远低于费米面(或价带)。
- 经典案例: 单质金属 Cu, 单质金属 Zn。
- PBE 的问题: PBE 确实仍然会把它们的 d¹⁰ 能带算得偏高(例如,对 Cu 金属,PBE 算出的 d 带顶距离费米面约 -2 eV,而实验值更低约 -2.5 eV)。
- 为什么可以不加 U?
- 在这些金属中,起决定性作用(如导电性、成键)的是 4s 电子,而不是深埋的 3d¹⁰ 电子。
- 虽然 d¹⁰ 能带的位置不完美,但这个“小错误”对晶格常数、体模量、费米面形状等关键物理性质的影响很小。
- 标准 PBE 已经能很好地描述这些金属的宏观性质,强行加 U 反而可能需要复杂的调参,甚至可能破坏原有的平衡。
结论:对于 3d¹⁰ 的金属单质,标准 PBE 计算通常被认为是“足够好”的基准。只有当你特别关心 d 带的精确位置(例如,用于模拟光电子能谱 XAS/XPS)或研究 d 带参与的精细化学过程(如表面催化)时,才需要考虑加 U 或使用更高级的泛函。
以 Ni(更典型的例子是 NiO)和 Zn(更典型的例子是 ZnO)来对比:
- NiO (Ni²⁺, 3d⁸): 3d 电子未占满。
- ZnO (Zn²⁺, 3d¹⁰): 3d 电子已占满。
核心区别:定性错误 vs. 定量错误
- NiO (d⁸, 未满): PBE 犯了“定性错误”
- 物理现实: NiO 是一个典型的莫特绝缘体 (Mott Insulator)。它的 d 电子由于强烈的在位库仑排斥(U)而高度局域化,导致其成为宽带隙绝缘体(实验带隙约 4.0 eV)。
- PBE 的计算结果: PBE (不加U) 无法处理这种强关联。它错误地将 d 电子描述为“巡游”的,导致能带重叠,错误地预测 NiO 是一个金属(没有带隙)。
- 加 U 的目的 (LDA+U): 在这里,U 是用来“惩罚”电子的巡游行为,**强迫 d 电子局域化**。这个 U 在自旋向上和自旋向下的能带之间“打开”了一个能隙(Mott-Hubbard 隙)。
- 总结: 加 U 是为了修正一个定性的、0 vs. 1 的错误(金属 vs. 绝缘体)。
- ZnO (d¹⁰, 全满): PBE 犯了“定量错误”
- 物理现实: ZnO 是一个常规的**带状绝缘体 (Band Insulator)**。它的 3d¹⁰ 轨道全满,深埋在价带(O 2p)之下。实验带隙约 3.4 eV。
- PBE 的计算结果: PBE (不加U) 正确地预测 ZnO 是一个绝缘体(因为 d 壳层全满了,它天然有带隙)。但是,PBE 的自相互作用误差导致 d¹⁰ 能带的能量位置算错了(算得太高),并与 O 2p 轨道产生了过度的虚假杂化,最终导致带隙被严重低估(算出来仅 0.8 eV 左右)。
- 加 U 的目的 (LDA+U): 在这里,d 电子已经是“局域”的(它们在全满的壳层里)。加 U 的目的不是为了打开能隙(能隙本来就有),而是为了“修正”d¹⁰ 能带的能量位置。U 将 d¹⁰ 能带推向更深的束缚能,解除了与 O 2p 的虚假杂化,从而“修正”了带隙大小,使其更接近实验值。
- 总结: 加 U 是为了修正一个定量的错误(带隙 0.8 eV vs. 3.4 eV)
ZnO 中当然有 d 电子的库仑排斥作用,而且非常强。
您的物理直觉是完全正确的。d 轨道的局域性是它的“本性”,这与它是否满占据无关。Zn 3d 轨道和 Ni 3d 轨道在空间上都是非常局域的,因此电子“挤”在同一个原子上的库仑排斥(U)都非常大。
那么,为什么我们在讨论 NiO 和 ZnO 时,对 U 的描述听起来如此不同呢?
关键区别在于:这个强大的“U”在 d 壳层“未满”和“全满”时,所导致的“物理后果”不同,因此我们使用 DFT+U 去“修正”的目标也不同。
1. NiO (d⁸, 未满): U 决定了“能不能动” (局域化)
- 物理情景:
- Ni²⁺ 的 d 壳层未满 (d⁸),这意味着 d 轨道上既有电子,也有空位。
- 理论上,一个 Ni 上的 d 电子可以“跳” (hopping) 到邻近 Ni 的 d 空位上,从而在晶体中巡游,形成能带,使材料导电(成为金属)。
- 但是,U 登场了。
- 这个“U”就是指当一个电子“跳”到邻近 Ni 上时,它必须与那个 Ni 上已有的 d 电子“共处一室”。这个新来的电子会受到其他 8 个电子的巨大库仑排斥。
- 这个能量代价(U)是如此之大,以至于电子“跳”不起来。
- 后果:
- 电子为了避免这个高昂的 U 代价,选择“放弃巡游”,老实地“局域”在自己的原子上。
- 这种由 U 引起的局域化,阻止了电子运动,从而在能带图上打开了一个能隙。这就是**莫特绝缘体 (Mott Insulator)**。
- PBE 的问题: PBE 低估了这个 U,错误地认为电子可以“跳”,所以算出来是金属。
- 加 U 的目的: 模拟这种局域化行为,强行“打开”这个Mott能隙。
2. ZnO (d¹⁰, 全满): U 决定了“有多深” (能级位置)
- 物理情景:
- Zn²⁺ 的 d 壳层已经全满了 (d¹⁰)。
- 关键点: 因为壳层全满了,d 电子没有空位可跳。它们天然就不能在 d 轨道之间巡游。它们本来就是局域化的。
- 所以,U 在这里不再扮演“阻止跳跃”的角色。
- 但是,U 依然存在。
- 这 10 个 d 电子之间强烈的内部库仑排斥(U),决定了这套 d¹⁰ 能带的绝对能量位置。
- 后果:
- 这套 d¹⁰ 能带是满的,它不是导带也不是价带,而是深埋在价带(O 2p)之下的“半芯态”。
- U 决定了这套 d¹⁰ 能带应该“埋”得多深。
- PBE 的问题: PBE 的自相互作用误差错误地估计了这套 d¹⁰ 能带的能量位置。它把这套 d¹⁰ 能带算得“太浅”了(能量过高,过于接近 O 2p 价带)。
- 加 U 的目的:
- 不是为了打开能隙(d 壳层全满,本身就不导电)。
- 而是为了“修正能级”。
- 加 U 之后,PBE 被迫承认 d 电子有更强的排斥,其效果是将这套 d¹⁰ 能带“推”回到更深、更物理的能量位置。
- 这消除了 d¹⁰ 能带与 O 2p 价带的虚假杂化,从而修正了价带顶的位置,最终修正了带隙大小。
但是Zr4+的3d电子是满的,那3d电子的库伦相互作用也很大,为什么不用考虑呢
您提出了一个非常敏锐且关键的物理问题!您的观察是正确的,这个问题的答案是区分“芯电子”和“价电子”的关键。
您的观察:Zr⁴⁺ (和 Zr 原子) 的 3d 电子确实是满的 (3d¹⁰)。 您的问题:为什么这个 3d¹⁰ 不需要加 U,而 Zn 的 3d¹⁰ 却需要?
答案是:因为 Zr 的 3d¹⁰ 是一个深能级的“芯能级” (Core Level),而 Zn 的 3d¹⁰ 是一个浅能级的“价能级” (Valence Level) 或“半芯能级” (Semi-core Level)。
这个能量位置的巨大差异,导致了它们在计算中截然不同的处理方式。
1. 能量位置的巨大差异 (物理现实)
- Zn (3d¹⁰) 在 ZnO 中:
- Zn 3d¹⁰ 能带的能量非常浅。
- 它位于 O 2p 价带的正下方(束缚能大约在 -7 到 -10 eV,非常接近价带顶 VBM)。
- 后果: 正是因为它离价带太近,PBE 算错它的位置(算得太高)时,它就会严重地与 O 2p 轨道发生虚假杂化,从而“污染”价带顶,导致带隙计算错误。
- Zr (3d¹⁰) 在 ZrO₂ 中:
- Zr 3d¹⁰ 能带的能量极其深。
- 它位于 O 2p 价带下方几百电子伏特 (eV) 的地方。(实验上,Zr 3d 的束缚能大约在 -180 eV 附近)。
- 后果: 这个能级离价带(约 0 eV)和导带(约 5-6 eV)太远了。PBE 就算算错了它的位置(比如算成了 -175 eV 或 -185 eV),这个“错误”也毫无意义,因为它绝对不会与 O 2p 价带或 Zr 4d 导带发生任何杂化。
总结: 我们为 Zn 3d 加 U,是因为它是一个“惹是生非”的浅能级,其位置错误会严重影响我们关心的带隙。 我们不为 Zr 3d 加 U,是因为它是一个“与世隔绝”的深能级,无论它在哪里,都不会影响我们关心的带隙。
2. VASP 赝势的处理 (计算现实)
基于上述的物理现实,VASP(以及所有赝势方法)在实际计算中会做如下处理:
- 赝势 (Pseudopotential) 的基本思想就是:只显式计算“价电子”,而把“芯电子”冻结在原子核周围,用一个势来等效替代。
- 对于 Zn (锌):
- 因为 3d¹⁰ 轨道能量很浅,会参与化学成键(或至少会干扰成键),所以它必须被当作“价电子”来显式计算。
- 因此,VASP 的 Zn 赝势(如
Zn_pv或Zn)会将 3d 和 4s 电子(共 12 个电子)都当作价电子。 - 既然 3d 被显式计算了,你就有机会 (也需要) 对它加 U。
- 对于 Zr (锆):
- 因为 3d¹⁰ 轨道能量非常深,是典型的芯电子,不参与化学成键。
- VASP 的 Zr 赝势(如
Zr_sv)会将 1s, 2s, 2p, 3s, 3p, 3d 轨道全部“冻结”在芯能级里,根本不显式计算它们。 - VASP 显式计算的“价电子”是 4s, 4p, 5s, 4d。
- 既然 Zr 3d 轨道从一开始就没被 VASP 算,你根本没有机会(也完全不需要)对它加 U。
一句话总结:
我们只对那些被 VASP 当作“价电子”来显式计算、且能量位置接近费米面(价带)的 d/f 轨道考虑加 U。Zr 的 3d 是深芯电子,不在此列。
重要
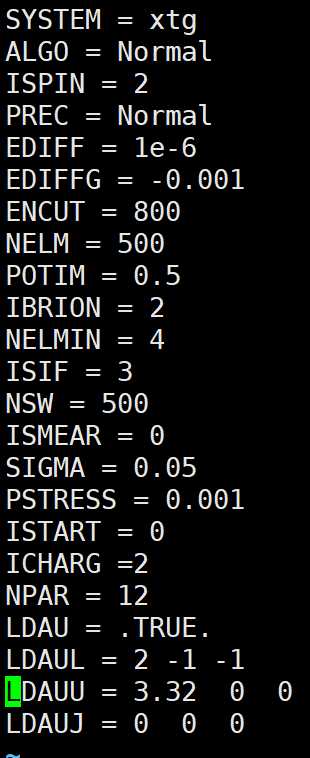
- 过渡金属元素需要加磁性ISPIN=2(可以不设置磁矩让它自己去优化磁矩),且算法不能为davision(normal),应该换成RMM(fast/veryfast),否则会电子步不收敛。可以不用加U,因为U是一个经验值,和实验来对的
加U无助于电子步收敛,加自旋有助于电子步收敛
可以都加上ISPIN=2 让它自动的去优化磁矩。
grep magnetization OUTCAR可以查看磁矩
关于结构优化中U和磁性
结构优化是根据能量进行结构的优化,使得能量向减小的方向演化,可能没有加U的能量不对,但是这和结构优化不收敛是不相干的。结构优化不收敛就是算法的问题。磁性还是有必要加的
DFT+U理论及“一步”完成DFT+U多步计算 - 知乎 (zhihu.com)
带你解读DFT+U计算,如何加U - 知乎 (zhihu.com)
重要
关于LASPH和LMAXMIX
vasp 4.6只计算PAW球内球形电子云的贡献,当轨道包括d,f等非球形电子云时,通过设置LASPH = True,可以考虑非球形电子云的贡献。
此外,当使用高级泛函 如 DFT + U,杂化泛函等时,考虑非球形电子云也能够计算更加精确的电子结构和能量。
vasp默认是关闭LASPH的
LMAXMIX控制直到哪个量子数范围内的PAW电荷密度会通过电荷密度混合器处理并写入CHGCAR文件。通常不需要增加LMAXMIX,但当进行DFT+U计算时,需要将LMAXMIX增加到4(对d电子体系)或6(对f电子体系)。对于DFT+U计算能带结构时,必须要提高LMAXMIX。考虑非共线磁性的自旋密度泛函理论(SDFT)时,需要考虑增加LMAXMIX
13. 磁性
通常磁矩是通过查看未成对电子数量来设置的,同时也要看轨道,对于弥散的轨道(s p),即便有未配对电子,电子的取向仍是随机的,而不是规则排序的。每个未成对电子对应最大磁矩为1
需要设置LORBIT = 11 然后在vasp的OSZICAR中,可以查看最终的总磁矩,在OUTCAR中,关键词mag可以查看分原子的磁矩。
磁矩通常是从上向下优化的,可以设置一个比较大的初始磁矩,让它从上向下优化
在一个磁性原子周围,可能会存在原本非磁性原子带有一些磁性的现象,这在计算中可以观察到
材料的磁性对应的微观电子结构为电子自旋的方向并不均匀分布。
绝大多数的磁性材料计算中,电子的自旋只会平行排列(磁各向同性,是共线磁矩
不考虑自旋的占据态能级自旋向上的能级和向下的能级相同,由同一个能级表示。
考虑共线磁矩时,自旋向上和自旋向下的能级分别输出,每个能级的电子占据数为1,共同计算费米能级。
由于自旋上和下能级大小不同,如果费米能级以下的不同自旋指标的能级数量不同,将导致体系整体出现磁矩。
对于反铁磁的情况,体系的磁密度呈现整体无磁矩,局部有磁矩的情况。
非共线磁矩
有的体系中,电子的自旋会沿不同方向排列(磁各向异性),这时磁密度通过x y z三个方向的投影表示
Davidson 算法对于磁性材料(ISPIN=2)有可能无法收敛,这时候解决的办法是更换电子算法为RMM (veryfast),或者手动设置磁矩 MAGMOM
14. 如何设置参数加快计算速度
- 注意kpar 和ncore的设置,**在计算带时不要跨节点计算!**(总核数/kpar/ncore 应该是节点数量的整数除数)
- LREAL 打开
- 减小KSPACING 和ENCUT
15. 关于vasp的输出中的free energy TOTEN 和energy without entropy 和 sigma >>0
在没有展宽时,(ismear = -5) 自由能和without能量是一致的,TOTEN和without的差异是展宽导致的
For the calculation of the total energy in bulk materials, we recommend the tetrahedron method with Blöchl corrections (ISMEAR=-5). This method also gives a good account of the electronic density of states (DOS). The only drawback is that the method is not variational with respect to the partial occupancies. Therefore the calculated forces and the stress tensor can be wrong by up to 5 to 10% for metals. Only for semiconductors and insulators, the forces are correct because the partial occupancies do not vary and are either zero or one.


12. 计算声子谱
一个技巧是可以用SPOSCAR做自洽算出CHGCAR和WAVECAR,用作其他微小位移的结构的初猜CHGCAR,在对微小位移结构做自洽时,需要设置ICHARG = 1 (读取但是会迭代)不能用ICHARG = 11 (读取后保持稳定不迭代)
1. 有限位移法 phonopy
生成超胞,自动搜索原胞
phonopy -d --dim 2 2 3 --pa auto生成力常数矩阵
phonopy -f disp-{001..003}/vasprun.xml计算dos,绘制pdos
phonopy-load --mesh 20 20 20 -p
phonopy-pdosplot -i '1 2 4 5, 3 6' -o 'pdos.pdf' partial_dos.dat
计算声子谱,得到band.yaml文件
phonopy-load --band auto -p绘制声子谱
phonopy-bandplot band.yaml -o band.jpg得到band.dat文件(可以直接用下面的python 脚本,更准确)
phonopy-bandplot --gnuplot band.yaml >> band.dat
2. 绘制声子谱python脚本
#处理band.yaml
#要求每一个路径的k点个数都相同,最好和phonopy官方的图对比一下
import yaml
import numpy as np
import matplotlib.pyplot as plt
import matplotlib.font_manager as fm
# --- 1. 加载 band.yaml 文件 ---
with open('band.yaml', 'r') as f:
data = yaml.safe_load(f)
# --- 2. 提取数据 ---
nqpoint = data['nqpoint']
nband = data['natom'] * 3
segments_nq = data['segment_nqpoint']
for i in range(1, len(segments_nq)):
if segments_nq[i] != segments_nq[0]:
print('Warning: 检测到不一致元素,需要修改脚本')
labels_list = data["labels"]
# 提取距离、频率和高对称点标签
distances = []
frequencies = []
for q_point in data['phonon']:
distances.append(q_point['distance'])
frequencies.append([band['frequency'] for band in q_point['band']])
distances = np.array(distances)
frequencies = np.array(frequencies)
# --- 3. 保存为数据文件
output_data = np.hstack((distances.reshape(-1, 1), frequencies))
# 保存为 band_py.dat,格式为:距离 Freq1 Freq2 Freq3 ...
np.savetxt(
'band.dat',
output_data,
fmt='%.8f',
header='Distance Frequencies(THz)...'
)
print("数据已成功提取并保存到 band_py.dat")
# print("高对称点位置:", labels)
n_path = len(labels_list)
n_points = nqpoint
n_per_path = n_points // n_path
# print(n_per_path)
gap_width = 0.02
x_prev = 0.0
new_distances = []
new_frequencies = []
xticks = []
xticklabels = []
"""
合并YAML格式的标签对,当相邻标签对满足前一个末端与后一个初段相同时合并
"""
merged_labels = [labels_list[0]]
segments_nq_new = [segments_nq[0]]
for i in range(1, len(labels_list)):
current = merged_labels[-1]
next_label = labels_list[i]
if current[-1] == next_label[0]:
merged_labels[-1] = current + next_label[1:]
segments_nq_new[-1] += segments_nq[i]
else:
merged_labels.append(next_label)
segments_nq_new.append(segments_nq[i])
if len(segments_nq_new) != len(merged_labels):
print('长度不匹配,有问题')
new_xticks = []
for i, label_path in enumerate(merged_labels):
start_idx = sum(segments_nq_new[:i])
end_idx = start_idx + segments_nq_new[i]
seg_dist = distances[start_idx:end_idx]
seg_freq = frequencies[start_idx:end_idx]
num_nodes = len(label_path)
points_per_subsegment = len(seg_dist) // (num_nodes - 1)
if i == 0:
seg_dist_shifted = seg_dist # 第一段无前置间隙
else:
seg_dist_shifted = seg_dist - seg_dist[0] + x_prev + gap_width # 加间隙
new_xticks.append(seg_dist_shifted[0])
new_xticks.append(seg_dist_shifted[-1])
new_distances.append(seg_dist_shifted)
new_frequencies.append(seg_freq)
# 记录高对称点位置和标签(保留间隙时,符号会重复显示)
# print(label_path)
for j in range(num_nodes):
if j == 0:
xticks.append(seg_dist_shifted[0])
else:
xticks.append(seg_dist_shifted[-1+ (j * points_per_subsegment)])
xticklabels.append(label_path[j])
# 更新上一段末尾位置(含当前段长度 + 间隙)
x_prev = seg_dist_shifted[-1]
# print(xticklabels)
# print(xticks)
font_path = '/usr/share/fonts/truetype/msttcorefonts/Times_New_Roman.ttf'
font_path_bold = '/usr/share/fonts/truetype/msttcorefonts/Times_New_Roman_Bold.ttf'
fm.fontManager.addfont(font_path)
fm.fontManager.addfont(font_path_bold)
plt.rcParams['font.family'] = 'Times New Roman'
plt.rcParams['axes.linewidth'] = 2
font_bold_prop = fm.FontProperties(family='Times New Roman', weight='bold')
plt.rcParams.update({
'mathtext.fontset': 'custom',
'mathtext.rm': 'Times New Roman',
'mathtext.bf': 'Times New Roman:bold',
'font.family': 'Times New Roman',
'font.weight': 'bold',
'axes.labelweight': 'bold',
})
fig = plt.figure(figsize=(8,6),dpi = 300)
# fig.subplots_adjust(left=0.2, right=0.8, top=0.8, bottom=0.2)
grid = fig.add_gridspec(1,1)
ax = fig.add_subplot(grid[0,0])
# print(len(new_distances))
for d, f in zip(new_distances, new_frequencies):
f = np.array(f)
for band in f.T:
ax.plot(d, band, color='red', linewidth=2)
ax.hlines(y=0, xmin=np.min(d), xmax=np.max(d), color='gray', linewidth=2, linestyle=':')
for tick in new_xticks:
plt.axvline(x=tick, color='black', linewidth=2, linestyle='-')
ax.set_ylabel(r"Frequency (THz)", fontsize = 35,fontname='Times New Roman',fontweight="bold")
xmin = min(arr.min() for arr in new_distances)
xmax = max(arr.max() for arr in new_distances)
ax.set_xlim(xmin, xmax)
ax.tick_params(
axis='x',
which='both',
top=False,
bottom = True,
labelbottom = True,
labeltop=False,
width=2
)
ax.tick_params(
axis='y',
which='both',
right=False,
left = True,
labelright=False,
labelleft = True,
width=2
)
plt.yticks(fontsize = 25,fontname = "Times New Roman",fontweight = 'bold')
ax.set_xticks(xticks)
ax.set_xticklabels(xticklabels,
fontsize=25,
fontname = "Times New Roman",
fontweight = 'bold')
plt.tight_layout()
plt.savefig('phonon_band_structure.png')
2. 关于并行参数
==解释==
第一性原理的计算过程是计算一个体系的不同k点上许多不同的电子态,不同的电子态也称为带/band,每个k点有很多要计算的带
假设一个场景,用一个cpu核计算一个体系,那么这个体系会先计算这个体系的第一个k点的第一个带,再计算第一个k点的第二个带,依次往后,计算第二个k点的第一个带。。。
并行指的是用多个cpu核同时计算多个任务。对一个计算而言,可以分为带并行和k点并行,也就是同时跑多少个k点,同时跑一个k点的多少个带。设置不同的并行参数,会分配给每个k点,每个k点的不同带cpu核心数。这两个并行并不是平行的,先对k点并行,然后给一个k点的带分配核数。
KPOINT 并行属于外层的并行,BAND 并行属于较内层的并行
比如一共有48核,一个k点要算的带数目为4,如果设置k点并行为6,带并行为4,那么就是同时计算6个k点,每个k点用48/6=8个核心来计算,同时计算一个k点的4个带,每个k点的每个带用2个核心计算。
如果一个k点要算的带数目一共只有3个呢,(当然最好设置带并行为3),假设设置带并行为2(设置为4会有一些核不进行计算),那么单个k点用48/6=8个核心,每个带用4个核心,同时计算2个带,但是这时候,会有一个带没有计算,在计算完一个带后,才会计算另一个k点,此时还会有一批cpu空下来,因此如果k点并行不设置为k点数目的除数,会出现空余的现象。 k点并行也一样,如果k点并行设置的太大,那么可能会有多的核数用不到
实际中应该把带并行和k点并行分开来看
一个重要的知识点
k点的并行优点是不需要通讯,因为没有数据的分发,缺点是要耗费内存。内存比较大的系统可以设置k点并行。
带的并行是把数据分发到不同的带上,这样的优点是耗费的内存少,缺点是不同带间需要通讯。当通讯带宽比较大的时候,可以把带的数目设置大一点,这样即便设置多个带并行通讯效率也不会太差;但是通讯带宽比较小时,不要设置同时并行太多带, 因为会大大增加带的通讯。
==cpu核数从哪里来?==
增加cpu的核数可以从两方面,一个是节点内并行,一个是多节点的并行。
节点内的并行相比节点间肯定是降低了通讯的时间的,但是有时候很难在一个节点内得到足够多的核心数,所以需要多个节点来串联计算。但是在设置并行参数时需要考虑的是尽量让k点和带的计算不要跨节点,比如给一个带分配50个核,但是一个节点只有48个核心,那么就需要去别的节点借来两个核心,由于节点间有通讯,这样会大大降低计算速度。不同节点的cpu核心串联时是依次串联的,因此可以先用第一个节点的,再用后面的。
也就是尽量保持节点间的独立性。
==vasp中的并行参数==
vasp中用KPAR来控制k点的并行,也就是一次计算多少个k点
KPAR默认为1,也就是并行计算一个k点,使用所有的核
vasp中用NPAR来控制带的并行,也就是每个k点同时计算多少条带
NPAR默认值为核的数量,比如有48核,NPAR默认为48,也就是同时计算48个带,每个带用1个核心计算,这一个核心计算完这一系列带再计算其他的带,其他的k点。
如果只设置NPAR,那么默认KPAR为1,NCORE=cpus/NPAR
带并行会把数据按照带来分发,这样需要增加不同带之间的通讯,当通讯带宽比较大的时候,可以把带的数目设置大一点,但是通讯带宽比较小时,不要设置同时并行太多带, 因为会大大增加带的通讯。
vasp中用NCORE来控制计算一个带的核心的数量(等同于NPAR的效果。 cpus/NCORE=NPAR)
NCORE 默认值为1 (与NPAR的默认值为所有的核数相对应,NCORE*NPAR的默认值为总核数)
==KPAR×NPAR×NCORE = cpus==
设置技巧
KPAR控制k点并行,这个参数应该设置为k点的除数,(不一定是k点本身的数目,这样就是同时算一个带的所有k点)
节点内并行要快于节点间的并行,因此尽量保证计算过程中一个节点计算整数条带,比如设置NCORE的值为一个节点的cpu核数,或者它的除数,这样保证一条带只在一个节点上计算
The best value NCORE depends somewhat on the number of atoms in the unit cell. Values around 4 are usually ideal for 100 atoms in the unit cell. For very large unit cells (more than 400 atoms) values around 12-16 are often optimal. If you run extensive simulations for similar systems, make your own tests.
让KPAR、NPAR、NCORE三个数比较相近
小体系并行
小体系的特点是原子数少, K点较多, 能带少. 因此KPAR应尽可能设大, NPAR可设为1.
在中等规模的系统(20-100 个原子)中,情况会有所不同。此时,计算的不可约k 点的数量很少(5-50),而电子能带的数量会很大(>100).
- 尽可能将 k 点并行化驱动到最大值(KPAR 尽可能大)。这里的限制因素是实际的 k 点数和可用的内存量。后者是因为更高的 KPAR 值会导致更高的内存需求。[2]
- 使用 Gamma 版本的 VASP 仅用于 Γ 点计算。它显着降低了内存使用量(3.7Gb→ 2.8Gb/core 用于 512 原子金刚石系统)并提高了计算效率,有时甚至提高了 2 倍。
- NPAR 并行化可用于减少高 KPAR 计算的内存负载,但就计算效率而言, 增加 KPAR 优于增加 NPAR。
- 如果只有 NPAR 并行化可用,由于 k 点太少,并且使用大型系统,NPAR 并行化也很重要。
K 点并行很重要!如果要大量计算类似结构, 如长时间的MD等, 应该在计算前简单测算选取最优的KPAR/NPAR(NCORE)参数.
NCORE 与 NPAR 只 能指定一个有效
NCORE = 8 下的运行时间经常处于最优值,且基本处于 [4,16] 空间范围内。
3. 关于K点
K-points里面的设置就是总共插入了多少个点,但是会由于对称性进行约化,得到的是不可约k点。比如 333出来14个k点,是因为约化了
http://bbs.keinsci.com/thread-13066-1-1.html
https://zhuanlan.zhihu.com/p/397873103?utm_id=0
关于测试K点和kSPACING
==需要保证能量差小于1mev/atom==
==k点没有想象中那么大,ENCUT没有想象中那么小==
先测ENCUT再测KSPACING
**金刚石测试结果(8 atoms) **PBE54
KSPACING = 0.1
KSPACING = 0.2
C的ENCUT=400 eV 用900才可以(注意是*100) KSPACING=0.1 2920 k点 KSPACING=0.2 365k点已经够了
PAW_PBE 0.2 (material project中使用的)

石墨测试(16atoms)

石墨的ENCUT需要用800 eV ,但是KSPACING可以用0.2
4.计算报错
试试ISYM = 0
1. internal ERROR RSPHER:running out of buffer
Abort(1) on node 15 (rank 15 in comm 0): application called MPI_Abort(MPI_COMM_WORLD, 1) - process 15
原因:buffer传输不够
解决方法:https://www.vasp.at/forum/viewtopic.php?t=2806&p=2809
改nonlr.F,重新编译vasp
search for the following lines in nonlr.F
IF (IRMAX > NONLR_S%IRMAX) THEN
! IRMAX is the maximum global number, could be improved !!!!
*NONLR_S%IRMAX =IRMAX 1.1
LREALLOCATE=.TRUE.
ENDIF
IF( IRALLOC > NONLR_S%IRALLOC) THEN
! more safety on parallel machines increase by 20 %
NONLR_S%IRALLOC =IRALLOC1.2*
LREALLOCATE=.TRUE.
ENDIF
and change
NONLR_S%IRALLOC =IRALLOC1.1 to
NONLR_S%IRALLOC =IRALLOC1.2
and
NONLR_S%IRALLOC =IRALLOC1.2 to
NONLR_S%IRALLOC =IRALLOC1.3
2 . 报错 Error EDDDAV: Call to ZHEGV failed. Returncode =
**1.NPAR **
2.赝势
**3.ALGO **
参见 http://www.error.wiki/Error_EDDDAV:_Call_to_ZHEGV_failed(在BN和NSmSiO自洽中遇到过)
3. 报错 ERROR FEXCP: supplied Exchange-correletion table is too small, maximal index :
编译器和vasp版本的冲突(编译的时候使用的是intel2018,使用时用intel2020会出现这样的问题),可能是POTCAR与vasp版本不兼容
vasp5.4-intel2020 vasp6.1-intel2018
一定要注意运行时使用的编译器版本和编译时使用的编译器版本相同(尤其是自己编译的)
4. ERROR FEXCP: supplied Exchange-correletion table is too small, maximal index : 3885
对称性的问题,设置ISYM = 0
5. Sub-Space-Matrix is not hermitian
我排查的问题是使用的节点太多了,减少使用的节点后可以运行
6. HNFORM: ERROR: k-point generating vectors and reciprocal lattice are incommensurate.
设置ISYM = 0
7.自洽不收敛
换电子算法
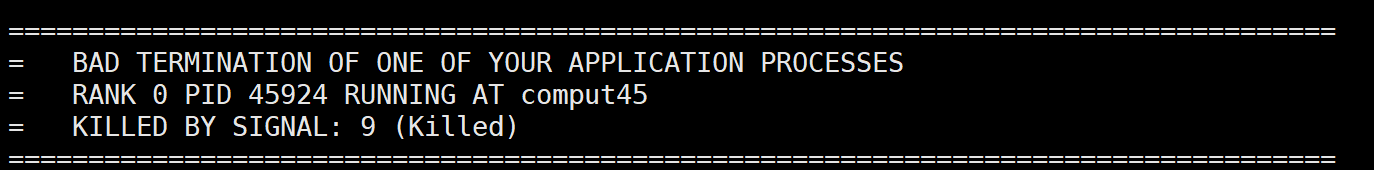
8. 内存溢出/BAD TERMINATION OF ONE OF YOUR APPLICATION PROCESSES
RANK 0 PID 45924 RUNNING AT comput45
KILLED BY SIGNAL: 9 (Killed)
内存的问题,设置的k点太密
9. 跑完第一个离子步报错 LAPACK: Routine ZPOTRF failed! INFO:1 KPOINT:1 SPIN:1
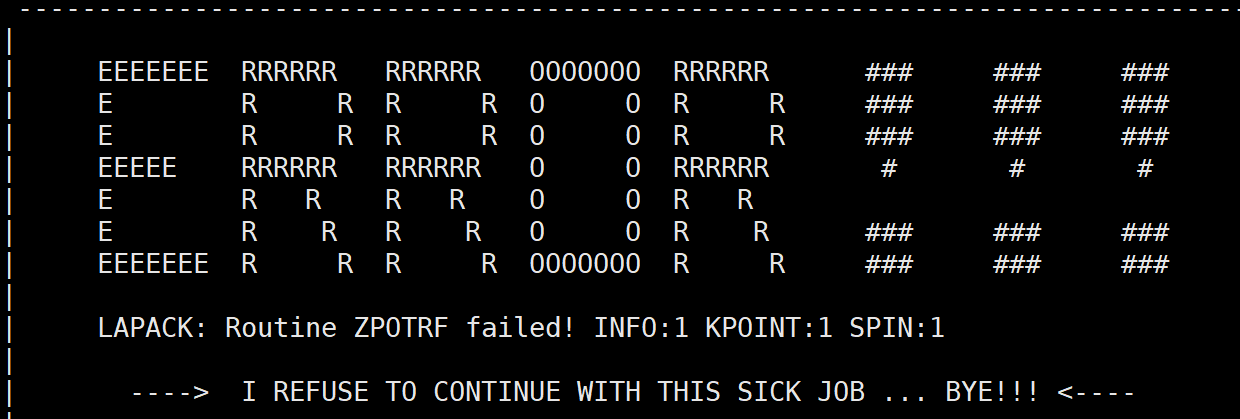
看看是不是在优化带真空层的结构,如果用了ISIF=3 会因为塌陷真空层太快而报错,可以改用ISIF=2或者ISIF=3调低 POTIM 然后循环优化
10. Intel MKL ERROR parameter 5 was incorrect on entry to DSTEIN2.
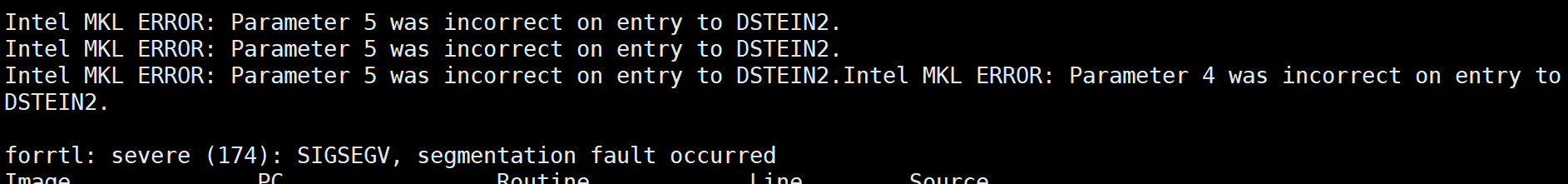
修改ALGO从Normal到Fast可以了
5.计算脚本
1.变压优化
for p in 100 200 300
#for i in $(seq 300 50 700)
do
cat > INCAR << lizhaohenshua
#cat 命令 查看文件的内容、连接文件、创建一个或多个文件和重定向输出到终端或文件 cat >test;cat a b > c; cat a;
#cat 后跟输入之后进行标准输出
#<<EOF是定界标识符 终结字符串必须写在行首 >是重写 >>是追加
SYSTEM=Li
PREC = Accurate
ENCUT = 600
EDIFF = 1e-6
IBRION = 2
ISIF = 3
NSW = 200
ISMEAR = 0
SIGMA =0.05
POTIM = 0.2
KSPACING = 0.18
EDIFFG = -0.001
lizhaohenshuai
mpirun -np 48 vasp路径 > vasp.log 2>&1``
cp CONTCAR CONTCAR_$p
cp OUTCAR OUTCAR _$p
done
6.tips
OUTCAR中的能量是晶格结构的能量,而OSZICAR中的能量E包括热浴和晶格结构的能量,OZICAR中的F是OUTCAR中的能量。按照经验来看,热浴的能量一直不平衡,但是系统的能量是平衡的。KPOINTS中,相同晶格常数应具有相同的k点密度,否则无法运行**vasp.log ! vasp.log! vasp.log!编译vasp:参考http://hmli.ustc.edu.cn/doc/app/vasp.5.4.1-vtst.htm
VASP 6.1.0 + VTST + intel新OneAPI 安装编译_vasp vtst onapi-CSDN博客
Installing VASP.6.X.X - VASP Wiki 可以多核并行来编译
- vasp的文件在桌面/vasp/vasp编译文件夹中
- source /work/env/intel2020
- 把makefile.include 复制到目录
- make all
*加背景电荷
NELECT= POTCAR中(ZVAL of the element) POSCAR中原子个数vasp
ALGO= All和ALGO = Fast/Normal的运行效果对比- 1.All的能量会略高于Fast 但是不多可忽略了
- 2.All花费的时间更长 Fast的单个电子步时长会慢慢变短 但是All维持一个时间长度。因此总的花费时间更长
- 3.All每个离子步中的电子步数大致相同,但是Fast会随着离子步增加收敛需要的电子步减少
7. 编译vasp
1.只编译vasp
把arch里对应的makefile.include复制到arch的上一文件夹
source intel环境

在makefile的文件夹下 make
注意:2025版的intel oneapi不支持ifort,icc,只有ifx icx,因此需要在makefile.include中把mpiifort改为mpiifx ,把icc改为icx 把icpx改为icx ,或者改icc为gcc icpx为g++
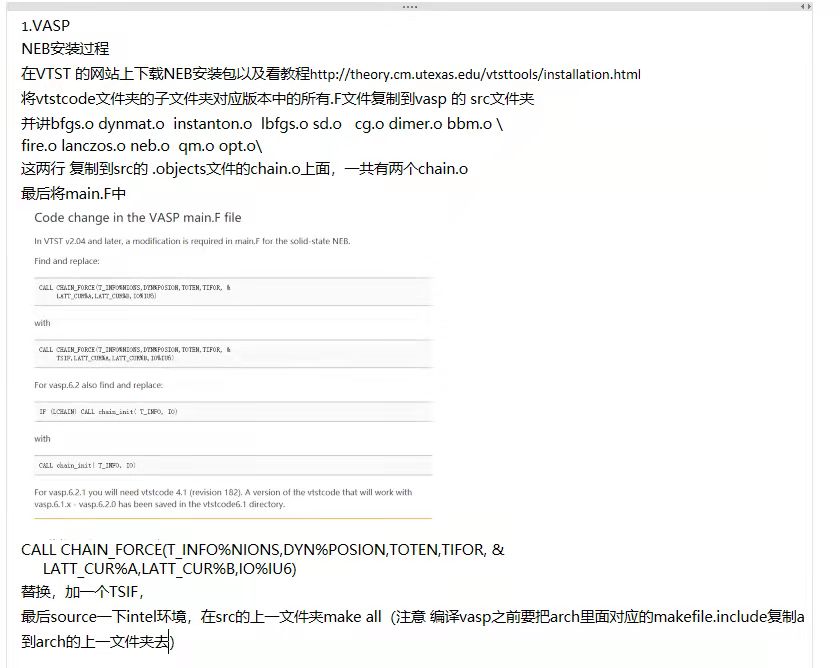
2.把vtst的neb方法编译进vasp
首先查看官方教程 vtst安装,官方网站给出了具体需要复制、修改哪些行
将vtstcode文件夹的子文件夹对应版本中的所有.F文件复制到vasp的src文件夹
将
bfgs.o dynmat.o instanton.o lbfgs.o sd.o cg.o dimer.o bbm.o \ fire.o lanczos.o neb.o qm.o opt.o \加入到vasp的src/.objects文件中,具体位置在chain.o之前。==一共有两个chain.o,都要复制上==

修改main.F

- source intel环境
- 复制对应版本的makefile.include
- make
原始版 教程
3. 编译可设置optcell的vasp

8. 使用MS技巧
如何去除原子周期性,这样删除一个原子不会删除所有等价位置的原子
build -symmetry-make p1
在做吸附时,如果发现复制一个分子上去会出现很多分子,这是由于主体结构包括对称性导致的,需要把主体结构make p1 然后再复制就会只出现一个分子
报错

原因:有道词典划词翻译? 关掉有道就好使了
9. 关于用零点振动能修正自由能
见模拟方法 OER台阶图
10. 关于vaspkit的KPOINTS和INPUT中的KSPACING的关系
vaspkit*2Π = KSPACING
因为vaspkit的单位是2Π/A ,而KSPACING的单位是1/A
vaspkit产生的KPOINTS可能会有错误,比如对真空层中一个水分子进行处理时,得到的KPOINTS是115这是不应该的
10.机器使用
申请192核然后用96核跑 scontrol show node nodelist 可能负载超过96,是因为开启多线程导致的,export OMP_NUM_THREADS=1用来控制关闭多线程
11. 生成正交包
转载请注明来源 有问题可通过github提交issue