1. 基本功能
Promoting transparency and reproducibility in enhanced molecular simulations
PLUMED中提供了各种分子动力学增强采样的方法,如umbrella sampling , metadynamics ,steered MD
这些增强采样方法需要和CV结合使用。CVs包括原子对之间的距离,扭转角,二次结构内容,回转半径,接触图,系统能量,与参考构想偏差的相关指标,基于人工网络人工定义的CVs
PLUMED可以用于on the fly 处理数据(感觉类似对msd的计算),也可用于增强采样
需要注意的是,PLUMED中的集体变量首先是表征系统的量,因此在给定集体变量或多集体变量后,可以按照一些函数输出对这个系统的刻画。如果需要用这些集体变量的函数值去bias系统,这是集体变量的额外功能,集体变量首先是静态的对系统的反映而不是动力学源头,因此首先把PLUMED当作数据分析软件学习,偏置是另一个大项
2. 使用lammps 调用plumed模拟
语法
fix ID group-ID plumed keyword value
keyword = plumedfile and outfile 这两个参量都是必需的
plumedfile PLUMED input
outfile PLUMED log 指定的outfile并不包括plumed的输出,而是输入文件的重复,plumed的输出文件在input文件中指定
fix plumed命令应该在时间步长等系统设置之后设置group-ID 不起作用,lammps 会向lammps传递所有原子信息
在输入文件中,只允许有一个
fix plumed命令,但是plumed本身允许多个独立集体变量,独立bias,和多种分析
example:
fix pl all plumed plumedfile plumed.dat outfile p.log
关于restart
注意需要在plumed的输入文件中开启plumed的restart
与plumed相关的热力学输出设置
fix_modify energy yes
输出plumed改变的势能面能量
fix_modify virial yes
输出plumed改变的压力
3. lammps + acnn + plumed 实现
plumd 只用来加速分子动力学,因此需要改变的地方只有lammps相关的
in.lmp中增加fix plumed命令fix pp all plumed plumedfile plumed.dat outfile p.log在
resource/md中增加plumed.dat文件
#目前采用的集体变量是氧原子的配位数
# Specify that we are in natural units
UNITS ENERGY=96.48530776074416 LENGTH=0.1
# Retrieve the value of the potential energy
ee: ENERGY
cn: COORDINATIONNUMBER SPECIES=61-132 R_0=5.0 MEAN
restraint: METAD ARG=cn.mean SIGMA=1 HEIGHT=0.5 PACE=10 FILE=HILLS
# Print the energy and the moduli of the three cell vectors
PRINT ARG=ee,cn.mean,restraint.bias FILE=colvar STRIDE=1
- 修改
resource/md/sub-md.sh中软件的路径,在安装时需要将plumed 静态链接到lammps中
目前在华为机器/home/jildxwlxyljlstdui/cccs-share02/lijx/liz/2-Na-X-Si-O/2_NaXSiO_big_model/1_one_element/14_Pr_es目录下的训练得到了验证
4. plumed 作为系统表征方法的输入文件
PLUMED的核心思想是计算可以描述变化的集体变量(collective variables),它们作为描述符刻画了系统发生的变化。对CVs的计算可以on-the-fly地进行,也可以对一段已有的轨迹后验地进行。
d:DISTANCE ATOMS=1,10
#d:命令代号 DISTANCE:命令名称 ATOMS:操作的原子
#计算1,10原子的距离
#标识符不能使用@开头,@在plumed有特定原子群
#所有的动作都可以被定义标识符并且引用
DISTANCE ATOMS=1,10 LABEL=d1
#同上,命令的另一种写法
phil:TORSION ATOMS=1,10,20,30
#计算1,10,20,30原子的扭转角
TORSION ...
LABEL=phi2
ATOMS=1,10,20,30
...
#一个分行来写的命令,计算扭转角
PRINT ARG=d FILE=COLCAR1 STRIDE=10
#在COLCAR1文件中每10步打印一次d的值。注意:只有在需要打印d的时候plumed才会计算一次d值
#STRIDE:默认值为1,只有在PRINT调用d时才计算一次d
PRINT ARG=phil FILE=COLCAR2 STRIDE=100
4.1 Collective Variables (colvar)
化学反应/物理过程的进行往往并不能通过单个原子的变化来观测;通过多个原子的协同变化能更好观测系统的变化,集体变量就是表征多个原子协同变化的量,如原子对距离、扭转角等。
这些集体变量可以直接输出,也可以作为函数的输入计算输出,也可以用于人为地偏置来加速动力学过程,也可以直接用来分析
构建集体变量的原则 (重要)
元动力学的基本思想是特定集体变量能够刻画系统的整体变化。偏置势是根据集体变量上定义的,作用是帮助系统在集体变量空间内更丰富地探索,推动系统在包括集体变量的整体势能面上探索。因此合适的集体变脸至关重要,需要用那些体现系统变化的重要特征
CV构建
选择原子集的方法:PLUMED: Groups and Virtual Atoms
CV变量表:PLUMED: CV Documentation
CV分析方法
CV打印方法:PLUMED: PRINT
CV的函数表:PLUMED: Functions
CV的偏置方法表:PLUMED: Bias
CV的分析方法表:PLUMED: Analysis
多CV的使用方法:PLUMED: MultiColvar
构建CV
1. 选择原子集
原子集的选择大部分是使用原子序号进行的。可以对特定的CV定义单独的原子集,也可以预先定义可重复使用的原子组。可以选定每个原子,也可以选定范围
g1:GROUP ATOMS=1,2,3,4,5 = GROUP ATOMS=1-5 LABEL=g1
GROUP ATOMS=10-50:10 LABEL=g3 = GROUP ATOMS=10,20,30,40,50, LABEL=g3
GROUP ATOMS=100-10:10 LABEL=g4
@mdatoms :MD模拟中所有的原子 ATOMS=@mdatoms
@allatoms :所有的原子,包括plumed中自定义的虚拟原子 ATOMS=@allatoms
#获得plumed中定义的虚拟原子
GROUP ATOMS=@allatoms REMOVE=@mdatoms
其他的选择原子快捷命令:PLUMED: MOLINFO
plumed可与MDAnalysis 和mdtraj联动选择原子
一些集体变量需要特定的原子数,distance 两个 ANGLE 3-4个 TORSION 4个原子
定义虚拟原子
CENTER 计算一组原子的几何质心
CENTER_OF_MULTICOLVAR 根据多集体变量计算一个加权平均的虚拟原子位置
COM 质心原子
FIXEDATOM 在特定的位置添加一个虚拟原子
GHOST 计算一个具有固定坐标的虚拟原子在三个原子构成的局部参考系中的绝对位置
引用虚拟原子
COM ATOMS=1,10 LABEL=com1
DISTANCE ATOMS=11,com1
监测一个虚拟原子:https://www.plumed.org/doc-v2.6/user-doc/html/_d_u_m_p_a_t_o_m_s.html
2. 选择CV函数
COORDINATIONc:COORDINATION GROUPA=1-30 R_0=0.3
特殊的CV -Disttance from reference configurations
distance from reference configurations对蛋白质折叠是一个很好的colvar,极大值和极小值分别对应蛋白质折叠和打开的状态。
直接用当前构型对应原子的位置减去参考构型的方法计算这一变量没有考虑质心平移和构型旋转。
RMSD是实际中计算这一描述符的方法。也可以计算多个与这一变量相关的集体变量来代替对真实的distance from reference configurations的描述
可用于计算distance from reference configurations的参数表:PLUMED: Distances from reference configurations
CV的函数
CV可以作为函数的输入计算函数的输出,用来表征结构。CV的函数需要使用PERIODIC 关键字来说明变量是否是周期性的。
CV函数表:PLUMED: Functions
4.2 MultiColvar/集体变量的分布
一个集体变量的值是对一组原子来计算的,多集体变量可以计算多组原子的某个集体变量的分布
当需要计算的并不是某个集体变量的值,也不是某个集体变量的函数的值(CV的函数),而是CV在构型中的分布时,需要用到multicolver。如计算两个原子最小的距离(需要计算所有的原子对距离),计算配位数大于3的原子。
$$
s=\sum _i g[f({X}_i)]
$$
$g[f(X_i)]$是集体变量f 的分布,Xi是不同的原子组
4.2.1 用法与输出
NAME <原子组> (<参数>) <计算的函数> TOL=0.001 LABEL=label
pos: GROUP ATOMS=220,221,235
neg: GROUP ATOMS=65,68,138,270,291
slt: DISTANCE GROUPA=pos GROUPB=neg
DUMPMULTICOLVAR DATA=slt FILE=MUTI.dat
如果需要输出f(Xi),需要用DUMPMULTICOLVAR命令 ,并且输出的标示符是DATA而不是ARG
4.2.2常用的multycolver
角分布函数;成键向量分布;桥连原子分布;原子配位数及其分布;密度;原子距离分布;氢键分布……
COORDINATIONNUMBER计算一组元素的配位数
4.2.3 多集体变量的函数 -筛选原子/计算函数值
有两种用来筛选原子的函数,一种是根据计算的多集体变量的值来筛选,如MFILTER_BETWEEN,MFILTER_LESS,MFILTER_MORE,另一种是只考虑特定区域内的原子(相当于先筛选再计算,如INENVELOPE (计算某种类型的原子密度比较高的区域内的集体变量的分布)后一种相当于一个$\delta $函数的权重
剩下还有一种函数用于计算多集体变量的值的函数如MTRANSFORM_BETWEEN
4.2.4 多集体变量的bias
系统的偏置就是对系统做限制,针对一些集体变量,使得系统的值保持在这个位置。比如固定质心、添加墙,同时保持分子动力学的正确性
4.3 特殊的集体变量-近邻矩阵
PLUMED可以计算$i_{th}$ 和$j_{th}$是否接触,构建接触矩阵
判断是否近邻有不同的方法,PLUMED提供了多种方法来判断是否近邻
4.4. PLUMED analysis 方法
PLUMED可以on the fly处理MD数据,也可以使用driver进行后处理
PLUMED分析方法:PLUMED: Analysis
输出
最简单的analysis是输出信息到文件中,PRINT和DUMPMULTICOLVAR都是analysis的方法
UPDATE_IF可以定义输出的范围,可以设置仅输出满足特定条件的量
COMMITTOR 定义满足条件时停止输出
STRIDE 定义计算并输出的间隔
重新加权和平均
REWEIGHT_BIAS 重新对系综平均加权以抵消偏置对相空间的影响
3. 其他输入参数
1. units
plumed有自己默认的单位制
Energy: kJ/molLength: nanometersTime: picoseconds
在plumed中,可以修改这些单位:LENGTH;ENERGY;TIME;MASS;CHARGE
Examples
UNITS LENGTH=A TIME=fs
UNITS LENGTH=0.1 TIME=0.001 #设置数字后单位与上面相等
5. PLUMED的增强采样-METAD
DP 运行实例,使用CP2K 模拟甘氨酸分子在纯水溶液中的质子转移
on-the-fly probability enhanced sampling
lammps官方例子 /home/share/jildxwlxyljlstdui/home/lijx/playground/acnn/interface/lammps/lammps-acnn/examples/PACKAGES/plumed
A quick introduction to PLUMED 2 (youtube.com)
Enhanced Sampling Methods - chapter 1: Free Energy and Sampling (youtube.com)
Meta dynamics Volume 1, September/October 2011
PLUMED中包含多种增强采样算法,METAD(元动力学只是其中一种)
1. 理论基础
元动力学偏置势的作用是将系统推离局域最小值。在长时间极限下,集体变量空间中的偏置势是集体变量真实势能面的负数
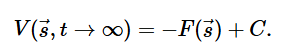
高斯函数的宽度决定了偏置势的平滑程度,高度决定了系统在该位置停留的时间
- 宽度太大,偏置势太平滑,导致出现徘徊;宽度太小,偏置势太尖锐,系统在集体空间中跳跃而不是平滑过渡
- 高度太小无法有效探索;高度太大过于迅速地掠过重要信息
高斯函数的信息输出到HILLS中,可以用于重启计算,也可根据CV重建自由能
在元动力学模拟中,随着模拟时间的增加,每一步需要评估的高斯核的数量增加,元动力学的成本也会增加。将偏置存储在网格上可以解决这一问题。
重启metadynamics的计算可以通过HILLS或者GRID进行。如果通过GRID进行,需要设置GRID_WFILE 来写入文件,并通过GRID_RFILE 来读取
关于well-tempered metadynamics
标准的元动力学中,每一次添加的高斯核的高度是固定的。在well-tempered metadynamics 中,高斯核的高度随着模拟的时间增加而减少, 偏置势能的收敛更加平稳
在长时间极限下,well–metadynamics的偏置势能与真实势能的关系是
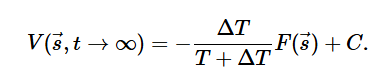
体系在高于设定温度$\delta T$ 的范围内采样 deltaT=0 表示标准MD,deltaT=无穷 表示标准的元动力学。通过设置deltaT的值决定在势能面探索的程度
bias factor (偏置因子)
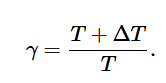
构建集体变量的原则 (重要)
元动力学的基本思想是特定集体变量能够刻画系统的整体变化。偏置势是根据集体变量上定义的,作用是帮助系统在集体变量空间内更丰富地探索,推动系统在包括集体变量的整体势能面上探索。因此合适的集体变脸至关重要,需要用那些体现系统变化的重要特征
机器学习势跑飞是因为一些原子跑到了未在第一性原理中探索的势能面,所以需要对跑飞时刻的原子构建集体变量,更广泛地探索这些原子的集体变量空间
from 智谱清言 (very useful)
在元动力学(Meta-Dynamics)中,集体变量(Collective Variables,CVs)是用于描述系统状态的一些选定的自由度,它们通常能够捕捉到系统动态中最重要的变化。集体变量可以是距离、角度、平面上的坐标、或是任何可以描述系统感兴趣特征的参数组合。
元动力学是一种计算方法,用于加速分子动力学模拟中稀有事件的采样。在元动力学中,为了促进系统探索其在集体变量定义的低能势面上的罕见区域,会在模拟中人为地添加一个外部势能,即所谓的“偏置势”。这个偏置势与系统的历史相关,意味着它会随着模拟的进行而不断更新。
这个偏置势是由一系列高斯函数(也称为高斯核)构建而成的,每一个高斯函数都对应于系统在其集体变量的历史位置。每当系统访问一个新的集体变量状态时,就会在这个状态上添加一个新的高斯偏置,这些新的高斯偏置使得新的集体变量的状态更容易到达和被采样。这些高斯函数的宽度决定了偏置势的平滑程度,它们的高度则与系统在该位置停留的时间成正比,或者是根据某种预定义的更新规则来调整。
通过这种方式,偏置势会逐渐在集体变量空间中形成一条势能降低的路径,引导系统从一个局部最小值移动到另一个局部最小值,从而促进那些在没有任何偏置时可能非常缓慢或不可能发生的转变。
在元动力学中,高斯核是构建外部历史相关偏置势的基本组成部分。每个高斯核都与系统在其集体变量(CVs)历史中的一个特定状态相对应。高斯核的具体作用可以从以下几个方面理解:
- 引导系统探索:高斯核的作用是引导系统在集体变量定义的低能势面上探索。通过在系统已经访问过的位置添加偏置势,元动力学鼓励系统探索新的区域,特别是那些能量较高、自然状态下难以到达的区域。
- 加速稀有事件的采样:在分子动力学模拟中,某些生物化学过程或物理转变可能非常缓慢,以至于在常规模拟时间内几乎不可能观察到。高斯核通过提供一条能量降低的路径,使得这些稀有事件的发生概率大大增加。
稀有事件的能量应该是不利的,通过偏置提供到达的路径
- 历史依赖性:高斯核的累积是历史依赖的,意味着偏置势随着模拟的进行而不断更新。这种历史依赖性确保了偏置势能够反映系统在集体变量空间中的完整历史,而不仅仅是当前状态。
- 平滑过渡:高斯函数的平滑性质使得偏置势在集体变量空间中形成连续的势能面,这有助于系统在势能面上平滑地过渡,而不是在离散的点之间跳跃。
- 自适应调整:高斯核的宽度和高度可以根据需要进行调整,以优化模拟效率和准确性。例如,较宽的高斯核可以提供更平滑的偏置势,而较窄的高斯核则可能导致更具体的引导。
- 避免局部最小值陷阱:在复杂系统的能量景观中,系统可能会陷入局部最小值,难以逃逸。高斯核通过在能量景观中构建“桥梁”,帮助系统跨越能垒,从而避免被困在局部最小值中。
总之,高斯核在元动力学中起着至关重要的作用,它们通过构建一个与系统历史相关的偏置势,引导系统在集体变量空间中高效地探索,从而加速对系统稀有事件的采样。
当然可以。让我们以蛋白质折叠过程为例来说明元动力学中高斯核的作用。
蛋白质折叠是一个复杂的过程,其中蛋白质从其未折叠状态转变到稳定的、功能性强的三维结构。这个过程中,蛋白质必须通过一个充满能量障碍的景观,这些障碍代表了中间状态,其中一些可能是亚稳态的,而另一些则可能是非功能性的。在没有任何外部帮助的情况下,蛋白质可能会在到达最终功能性结构之前陷入这些亚稳态。
在元动力学的框架下,我们可以选择一些集体变量来描述蛋白质的结构状态,比如两个特定氨基酸之间的距离、某些原子的角度或者是蛋白质的整体扭转。这些集体变量定义了蛋白质折叠的简化空间,我们可以在其中应用偏置势。
现在,让我们考虑一个具体的例子:
- 选择集体变量:我们选择蛋白质中两个关键氨基酸之间的距离作为集体变量。这个距离可以很好地描述蛋白质折叠过程中的关键转变。
- 初始化模拟:开始时,蛋白质处于未折叠状态,我们在其集体变量的当前值处放置一个高斯偏置势。这个高斯函数有一个特定的宽度和高度,决定了偏置势的形状和强度。
- 模拟过程:随着模拟的进行,蛋白质开始在集体变量空间中移动。每当蛋白质访问一个新的集体变量状态时,我们就在这个状态上添加一个新的高斯偏置势。随着时间的推移,这些高斯函数累加起来,形成了一个偏置势能面。
- 引导折叠:偏置势能面开始引导蛋白质沿着集体变量空间中的能量降低路径移动。这有助于蛋白质跨越能量障碍,避免陷入亚稳态,并最终达到低能的折叠状态。
- 更新偏置势:随着蛋白质继续移动,我们会根据预定义的规则定期更新高斯偏置势的参数,比如增加高度或改变宽度,以确保偏置势能够有效地引导蛋白质折叠。
通过这种方式,元动力学中的高斯核帮助我们模拟并理解蛋白质折叠的复杂过程,这在没有偏置的情况下可能需要非常长的模拟时间才能观察到。
在元动力学中,调整高斯核的参数对于获得有效的模拟结果至关重要。高斯核的主要参数包括宽度(σ)和高度(V),这些参数的调整会影响偏置势的形状和强度,从而影响模拟的效率和准确性。以下是一些调整高斯核参数的策略:
- 选择合适的集体变量(CVs):在添加高斯偏置之前,首先要确保选择了合适的集体变量,这些变量应该能够有效地描述系统的关键转变。如果集体变量选择不当,即使高斯核参数调整得很好,也可能无法获得满意的模拟结果。
- 调整宽度(σ):
- σ太小:偏置势会非常尖锐,可能导致系统在集体变量空间中跳跃,而不是平滑地过渡,这可能会增加模拟的噪声并降低效率。
- σ太大:偏置势会过于平滑,可能会导致系统在集体变量空间中徘徊,而不是有效地探索新的区域。
- 调整高度(V):
- V太小:偏置作用不足以有效引导系统探索新的区域,特别是对于能量较高的区域。
- V太大:可能会导致系统过于迅速地通过某些重要区域,从而错过关键过程或过度偏置,导致模拟结果失真。
- 自适应调整:可以设计算法,根据系统的进展自动调整高斯核的参数。例如,如果系统在某个区域停滞不前,可以增加偏置的高度来帮助系统跨越能量障碍。
- 平衡探索与利用:在元动力学中,需要平衡对已知区域的深入探索和对未知区域的大胆探索。可以通过调整高斯核的参数来实现这种平衡,例如,通过降低高度或增加宽度来鼓励探索。
- 使用交叉验证:可以通过在不同模拟中尝试不同的参数设置,并比较结果的一致性和准确性来优化参数。
- 考虑系统的动态特性:系统的动态特性,如能量障碍的高度和宽度,以及系统的自然时间尺度,都会影响最佳的高斯核参数选择。
- 实验设计:可以先进行一系列预模拟,以快速确定参数的大致范围,然后在这些参数的基础上进行更精细的调整。
总之,调整高斯核参数是一个迭代的过程,可能需要根据模拟的具体情况进行多次尝试和优化。通过仔细选择和调整这些参数,可以显著提高元动力学模拟的效率和有效性。
元动力学中引入的偏置势能够不断地对系统施加力,使其不停留在已探索过的局部能量最低点,而是继续向相空间中未知的区域移动。这样,系统就有机会访问到在自然条件下很难到达的状态,从而加快了对系统动态行为的全面理解。
随着时间的推移,偏置势的效果累积起来,最终反映出了系统在集体变量定义的低维空间中的自由能表面。在理想情况下,当模拟时间足够长时,偏置势会变得越来越接近于系统的真实自由能表面,不过是以相反的符号表示。这意味着偏置势的最高点对应于系统自由能的最低点,反之亦然。因此,通过分析偏置势的形状,我们可以得到系统自由能景观的轮廓,从而推断出系统在不同状态之间的转换路径和能量障碍。
元动力学(Metadynamics)是一种计算模拟方法,用于加速分子动力学(MD)中对稀有事件的采样。这个过程通常包括以下几个步骤:
- 选择集体变量(Collective Variables, CVs):首先,需要选择一组能够描述系统状态的参数作为集体变量。这些变量应该能够捕捉到系统动态中最重要的变化,例如蛋白质结构的变化、化学反应的进度等。
- 初始化模拟:开始时,系统处于初始状态,通常是一个能量较低的局部最小值。在集体变量的当前值处放置第一个高斯偏置势。
- 进行分子动力学模拟:在模拟过程中,系统会在原有的物理势能基础上,受到添加的高斯偏置势的影响。这个偏置势会随着模拟的进行而不断更新。
- 添加高斯偏置:每当系统访问一个新的集体变量状态时,就在这个状态上添加一个新的高斯偏置。这些高斯函数的宽度决定了偏置势的平滑程度,它们的高度则与系统在该位置停留的时间成正比,或者是根据某种预定义的更新规则来调整。
- 更新偏置势:随着模拟的进行,系统会不断地在集体变量空间中移动,高斯偏置势会根据系统的历史位置逐渐累积。这个累积的偏置势会逐渐在集体变量空间中形成一条势能降低的路径,引导系统从一个局部最小值移动到另一个局部最小值。
- 分析结果:模拟结束后,分析累积的偏置势可以得到系统的自由能景观。这个景观揭示了系统在不同状态之间的能量关系,以及转换路径和能量障碍。
- 优化参数(可选):根据模拟结果,可以调整集体变量和高斯偏置的参数,以提高模拟的效率和准确性。这可能需要多次迭代和模拟。
通过这个过程,元动力学能够帮助研究人员探索系统的能量景观,特别是在研究稀有事件,如蛋白质折叠、化学反应的过渡态等过程中非常有用。
偏置势势能面(Bias Potential Energy Surface)是在分子动力学模拟中,特别是在元动力学(Metadynamics)模拟中人为引入的一个额外的势能面。这个势能面是由一系列在模拟过程中动态添加的高斯函数(或其他形式的偏置函数)组成的,其目的是为了加速系统对相空间中的特定区域的探索,尤其是那些在自然条件下难以访问的区域。
在传统的分子动力学模拟中,系统遵循由原子间相互作用决定的物理势能面。然而,对于某些生物学或化学过程,如蛋白质折叠、分子对接、化学反应等,感兴趣的过程可能非常缓慢,以至于在可接受的模拟时间内很难观察到。这是因为这些过程涉及到必须跨越的高能垒。
为了克服这个问题,元动力学在物理势能面上叠加了一个偏置势势能面。这个偏置势随着模拟的进行而不断调整,其作用是“推动”系统探索新的区域,从而加速稀有事件的采样。偏置势的引入不会改变系统的物理性质,但它可以改变系统在相空间中的动态行为,使得原本缓慢的过程变得可行。
随着时间的推移,偏置势势能面逐渐接近于系统的自由能面。在长时间尺度下,偏置势势能面收敛到自由能面的负值,这意味着偏置势的最高点对应于自由能的最低点,反之亦然。因此,通过分析偏置势势能面的形状,可以获得系统自由能景观的轮廓,从而推断出系统在不同状态之间的转换路径和能量障碍。
在元动力学模拟中,GRID_SPACING 和 GRID_BIN 是两个用于定义如何将偏置势能(bias potential)离散化到网格上的参数。这两个参数的作用和设置方法如下:
- GRID_SPACING:这个参数定义了网格点之间的间距。在元动力学模拟中,偏置势能是由一系列高斯函数组成的,这些函数随着模拟的进行而不断添加。如果使用
GRID_SPACING,您可以直接指定网格点之间的距离。这个间距决定了网格的精细程度,间距越小,网格越细,模拟的精度可能越高,但计算成本也越大。 - GRID_BIN:这个参数定义了每个集体变量(collective variable, CV)方向上应该有多少个网格箱(bins)。网格箱的数量越多,网格越细,模拟的精度可能越高,但同样,计算成本也越大。
设置这两个参数时,应该考虑以下几点:
- 系统的复杂性:如果系统的能量景观非常复杂,或者集体变量的变化范围很大,可能需要更细的网格来准确捕捉能量景观的细节。
- 计算资源:更细的网格意味着更高的计算成本。您需要根据自己的计算资源(如CPU时间、内存等)来平衡网格的精细程度和计算效率。
- 模拟目的:如果您对模拟结果的精度要求非常高,可能需要使用更细的网格。但如果只是进行初步的探索,较粗的网格可能就足够了。
- 默认设置:如果既没有指定
GRID_SPACING也没有指定GRID_BIN,并且高斯宽度是固定的,PLUMED会默认使用高斯宽度的1/5作为网格间距。这是一个经验性的合理选择,但对于特定的系统可能需要调整。
在实际操作中,您可以先从默认设置或根据经验选择一个初始值,然后进行测试模拟,观察结果是否稳定和准确。如果发现结果不够理想,可以尝试调整网格参数,进行新一轮的模拟,直到得到满意的结果。
2. PLUMED中的METDA参数
必填关键词
SIGMA: 高斯势的宽度PACE: 增加势的频率FILE: 增加的势存储的位置HEIGHT高斯势的高度
选填关键词
GRID_BIN/GRID_SPACING储存高斯函数的网格的精细度,SPACING越小,网格越细,模拟的精度越高,计算量越大。默认值为1/5的高斯宽度GRID_WFILE and GRID_WSTRIDE)写网格文件,可以用于重启计算,也可以用HILLS重启BIASFACTOR偏置因子,在优化的元动力学中使用,决定偏置的程度/势能面探索的程度TEMP系统温度,在优化的元动力学中使用
默认输出
bias: bias potential的瞬时值
work: 工作蓄能器
例子
例子:使用两对原子的距离做metadynamics
DISTANCE ATOMS=3,5 LABEL=d1
DISTANCE ATOMS=2,4 LABEL=d2
METAD ...
ARG=d1,d2 SIGMA=0.2,0.2 HEIGH=0.3 PACE=500 LABEL=restraint
...
PRINT ...
ARG=d1,d2,restraint.bias STRIDE=100 FILE=COLVAR
...
phi: TORSION ATOMS=__FILL__
# Compute the backbone dihedral angle psi, defined by atoms N-CA-C-N
psi: TORSION ATOMS=__FILL__
# Activate well-tempered metadynamics in phi
metad: __FILL__ ARG=__FILL__ ...
# Deposit a Gaussian every 500 time steps, with initial height equal to 1.2 kJ/mol
PACE=500 HEIGHT=1.2
# The bias factor should be wisely chosen
BIASFACTOR=__FILL__
# Gaussian width (sigma) should be chosen based on CV fluctuation in unbiased run
SIGMA=__FILL__
# Gaussians will be written to file and also stored on grid
FILE=HILLS GRID_MIN=-pi GRID_MAX=pi
...
# Print both collective variables on COLVAR file every 10 steps
PRINT ARG=__FILL__ FILE=COLVAR STRIDE=__FILL__
高斯势的高度是否下降为0不是判断元动力学是否收敛的参数
转载请注明来源 有问题可通过github提交issue